1. Оптимизация структуры порфирина с помощью программы Mopac
В файле 1.smi находится аннотация порфирина в виде SMILES. С помощью программы obgen построим его 3D струкртуру:
obgen 1.smi > 1.mol
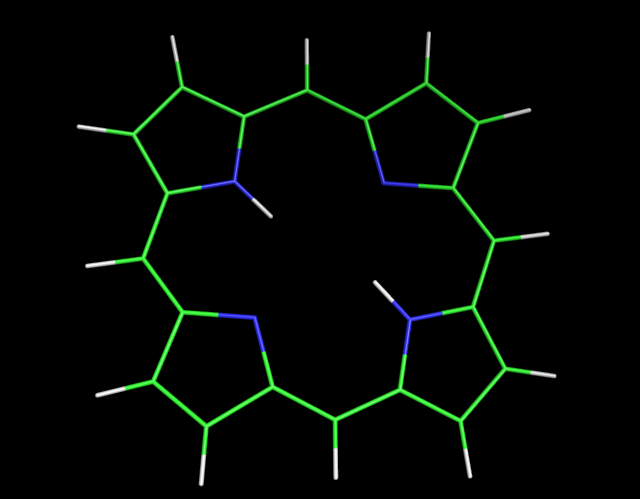
Просмотрим полученную структуру в PyMol и удалим ненужные водороды, сохраним результат в файле porphyrin.pdb .
|
|
Молекула не является плоской, хотя должна быть таовой.
С помощью babel переформатируем координаты в mol формате во входной файл для Mopac:
babel -ipdb porphyrin.pdb -omop 1_opt.mop -xk "PM6"
Полученный файл 1_opt.mop подоадим на вход программе Mopac:
MOPAC2009.exe 1_opt.mop
Переформатируем файл 1_opt.out в pdb:
babel -imopout 1_opt.out -opdb 1_opt.pdb
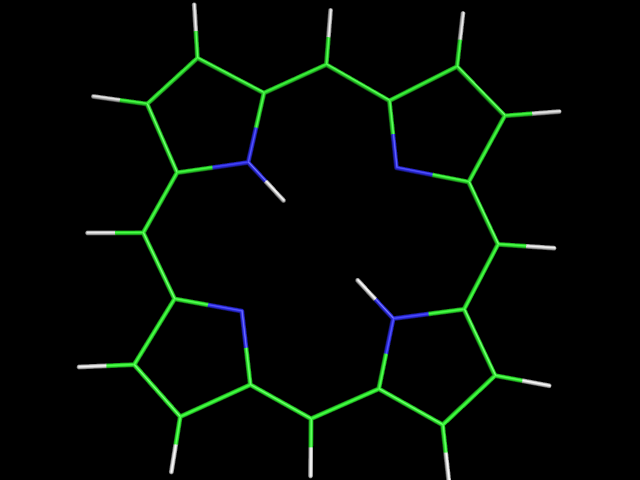
Посмотри на результат оптимизации в PyMol:
 |
 |
Теперь структара плоская.
2. Расчет возбужденных состояний порфирина и спектра поглощения молекулы
Для указания Mopac о необходимости расчёта возбуждённого состояния добавьте в конец файла 1_opt.mop пустую строку и строку "cis c.i.=4 meci oldgeo", назовем полученный файл 1_opt_spectr.mop .
Запустим MOPAC:
MOPAC2009.exe 1_opt_spectr.mop
Полученный файл 1_opt_spectr.out содержит значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
The "+" symbol indicates the root used.
По формуле E=hv, где h - постоянная Планка, а v - частота волны, найдем длины волн, соответсвующие электронным переходам:
E (eV) Длина волны (nm) 1.913312 649 2.266014 548 2.463186 504 2.823915 440 3.362161 369 3.389757 366 3.669242 338 3.871323 321
3. Оптимизация структуры парабензохинона
В файле 3.smi находится аннотация парабензохинона в виде SMILES.
Результат программы obgen в файле 3.pdb :
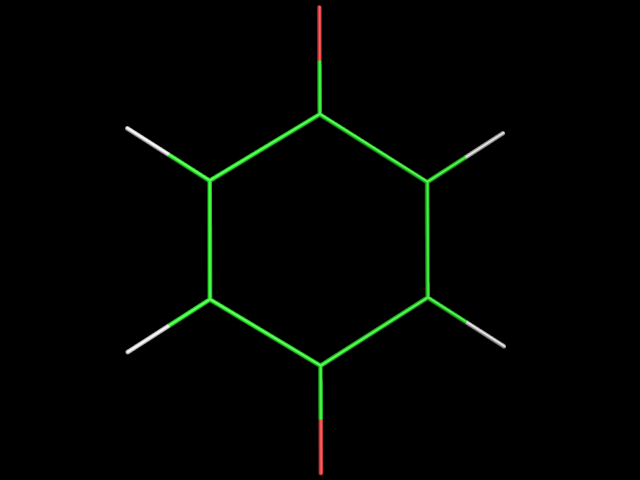
Результат программы Mopac в файле 3_opt.pdb :
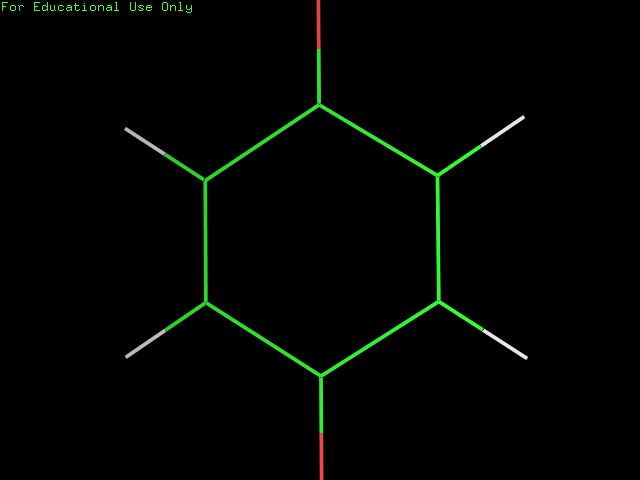
Полученные структуры выглядят очень похожими.
Определите геометрию дианиона этой молекулы, для этого в первую строчку mop файла добавьте слово CHARGE=-2, затем укажем, что заряд должен находится на атомах кислорода ( 3_2_opt.mop ).
На рисунке изображена структура парабензохинона (углеродные атомы синие), наложенная на структуру дианиона (углеродные атомы зеленые):
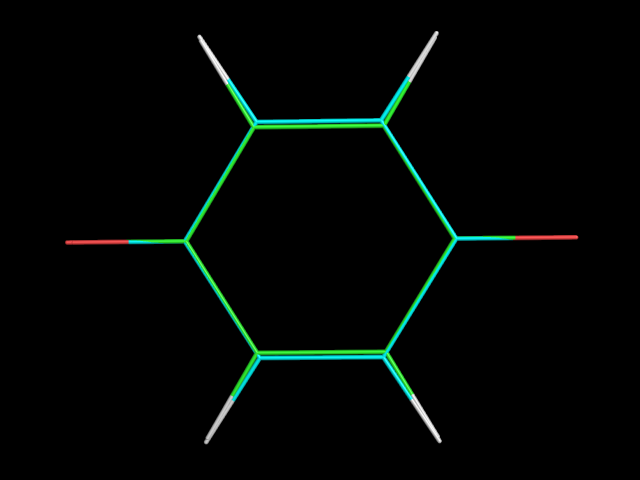
Кольцо дианиона немного уже, вследствие отталкивания отрицательно заряженных атомов кислорода.