1. Оптимизация структур азулена и нафталина с помощью программы Mopac
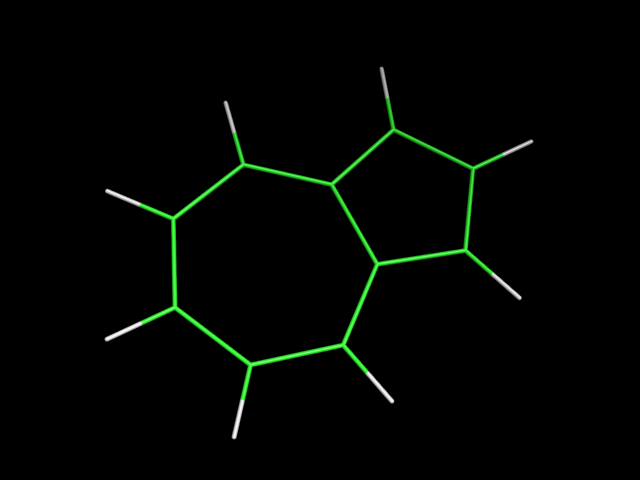
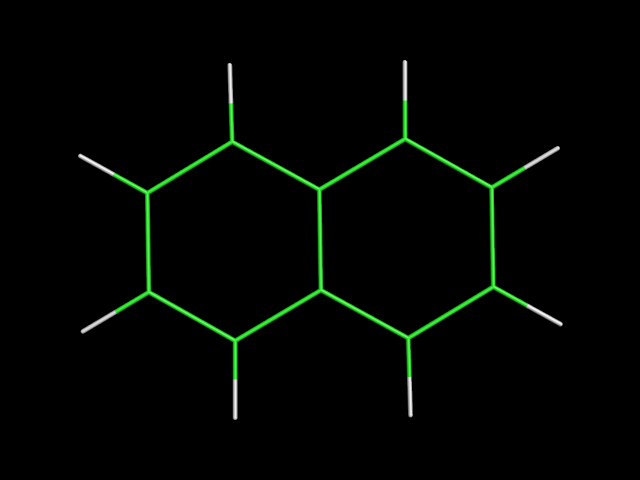
В файлах azulene.smi и napthalene.smi сохранены SMILES представления для азулена и нафталена. С помощью obgen получена структура молекул:
obgen azulene.smi -ff UFF > azulene.mol
obgen napthalene.smi -ff UFF > napthalene.mol
| азулен | нафтален |
| | |
Затем структуры сохранили как pdb файлы, с помощью babel переформатировали координаты в mol формат, запустили Mopac. В результате для азулена получили файл azu.out , для нафталена - nap.out .
С помощью babel переформатировали файлы в формат gamin:
babel -imopout azu.out -ogamin azu_opt.inp
babel -imopout nap.out -ogamin nap_opt.inp
Сделаем так, чтобы заголовки файлов выглядили так:
$CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=OPTIMIZE $END $BASIS GBASIS=N31 NGAUSS=6 $end $system mwords=2 $end $DATA
Полученные .inp файлы - входные для программы GAMESS.
2. Оптимизация геометрии с помощью GAMESS
Для оптимизации запустим Gamess:
gms nap_opt.inp 1 >& nap_opt.log gms azu_opt.inp 1 >& azu_opt.log
Оптимизация геометрии проводилась с базисом: 6 гауссовых функций для невалентных электронов и 4 гауссовые функции (2 блока: 3 и 1) для валентных электронов.
3. Расчет энергии методами Хартри-Фока и теории функционала плотности
На основе полученных координат составим новые входные файлы для расчёта энергии. Теперь построим по два файла на каждую молекулу, в первом случае будем вести расчёт методом Хартри-Фока, а во втором - используя теорию функционала плотности. Используя babel, переформатируем log файлы gamout в gamin и сохраним их в файлах nap_opt_2.inp и azu_opt_2.inp .
Для расчёта по Хартри-Фоку составим файлы с таким заголовком (в файлах nap_opt_2_HF.inp и azu_opt_2_HF.inp ):
$CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=ENERGY $END $BASIS GBASIS=N31 NGAUSS=6 $SCF DIRSCF=.true. $end POLAR=POPN31 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $system mwords=2 $end $DATA
В случае теории функционала плотности заголовок будет таким (в файлах nap_opt_2_DFT.inp и azu_opt_2_DFT.inp ):
$CONTRL COORD=CART UNITS=ANGS dfttyp=b3lyp RUNTYP=ENERGY $END $BASIS GBASIS=N31 NGAUSS=6 POLAR=POPN31 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $system mwords=2 $end $DATA
Результаты расчетов для азулена: azu_opt_2_HF.log (методом Хартри-Фока) и azu_opt_2_DFT.log (по теории функционала плотности).
Результаты расчетов для нафталена: nap_opt_2_HF.log (методом Хартри-Фока) и nap_opt_2_DFT.log (по теории функционала плотности).
4. Расчет энергии изомеризации
| Метод | Napthalene | Azulene | Δ, Hartree | Δ, kCal/mol* |
| Хартри-Фок | -383.355 | -383.282 | 0.072 | 45.301 |
| DFT | -385.640 | -385.586 | 0.054 | 34.05 |
* 1 Хартри = 627.509469 ккал/моль.
Эксперементально установленная энергия изомеризации нафталена в азулен составляет 35.3±2.2 kCal/mol. Значит результат, полученный с помощью теории функционала плотности (DFT) ближе к действительности.