Анализ качества прочтений
| fastqc chr3.fastq | Анализ качества прочтений |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr3.fastq chr3.trimmed.fastq TRAILING:20 MINLEN:50 | Фильтрация качеств ридов |
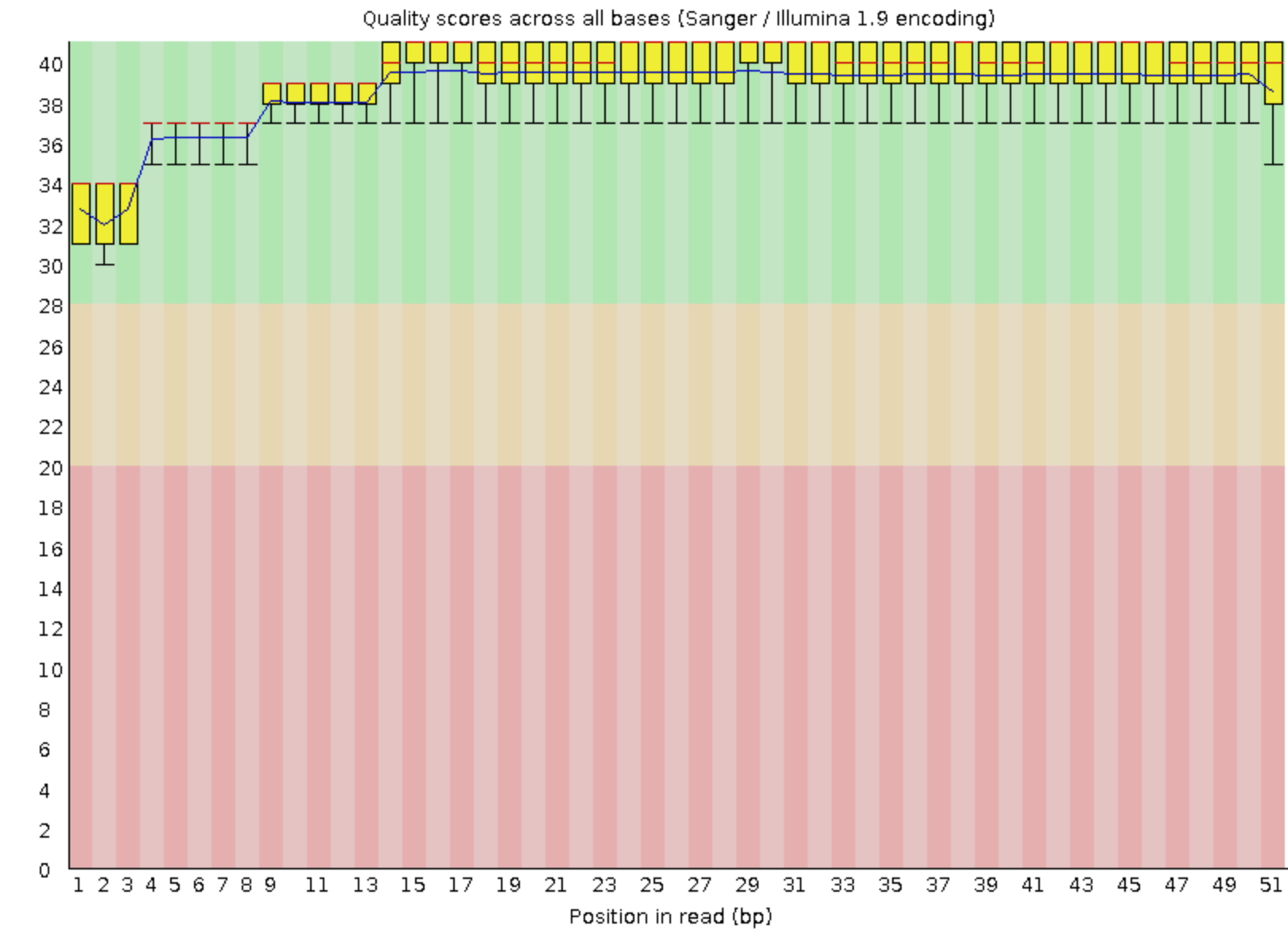
Видно, что качество хорошее, даже нет выбросов.
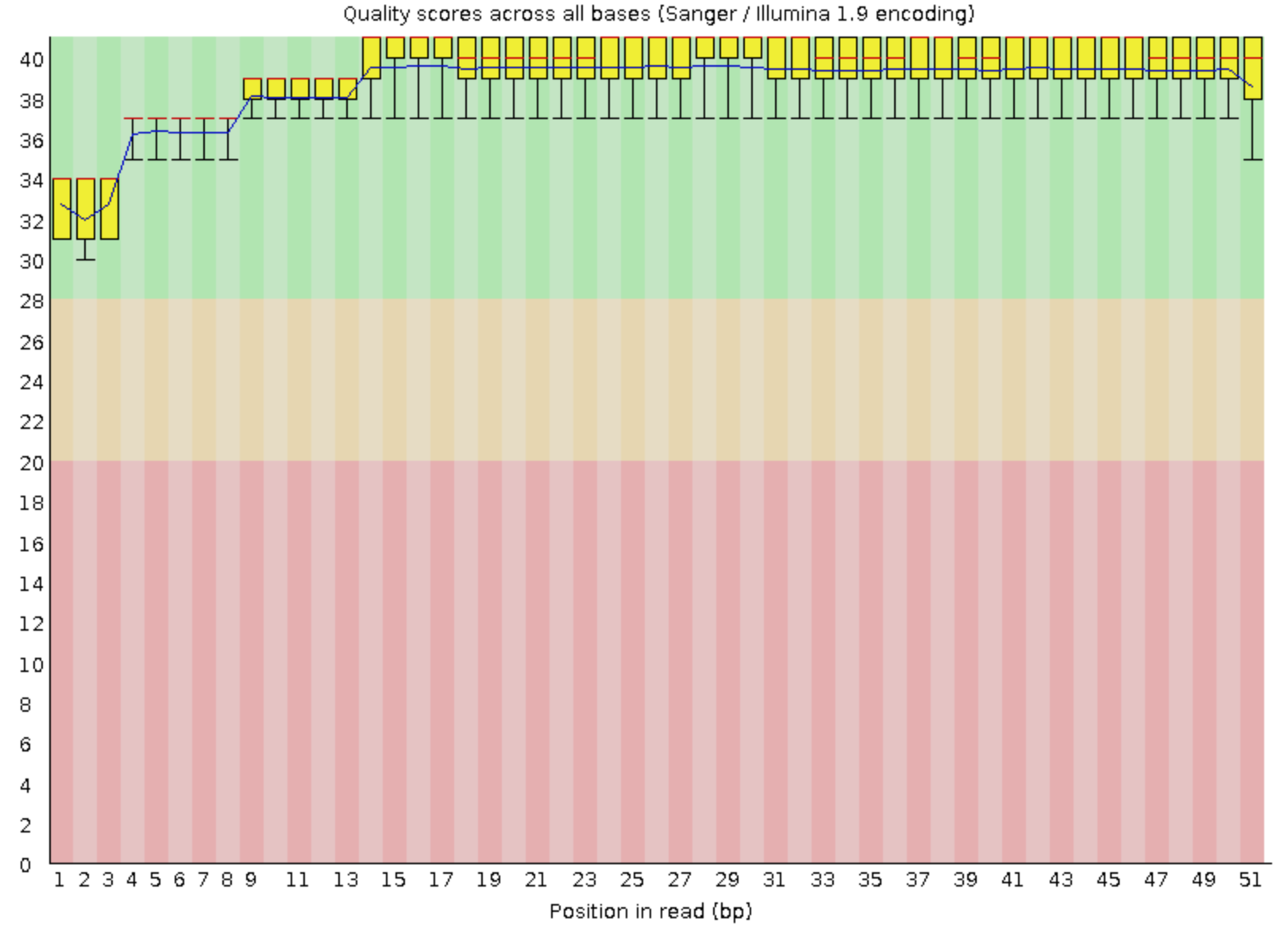
Выброшены были 59 последовательностей.
Анализ качества прочтений
| hisat2 -x chr3 -U chr3.trimmed.fastq --no-softclip > chr3.sam | Картирование прочтений, нужно убрать опцию --no-spliced-alignment, потому что как раз РНК может быть сплайсировано. |
| samtools view chr3.sam -bo aligned.bam | Переход в бинарный формат |
| samtools sort aligned.bam -T aligned -o chr3.sorted.bam | Сортировка bam файлов |
| samtools flagstat chr3.sorted.bam > flagstat.out | Статистика по картированию |
Накартировано было 21019 ридов, это 99.27% – это хорошо.
Подсчет чтений
| htseq-count -f bam chr3.sorted.bam -i gene_id -s no gencode.v19.chr_patch_hapl_scaff.annotation.gtf >chr3.counts | Подсчет количества ридов, отнесенных к каждому гену. |
| grep -wv 0 chr3.counts | Поиск ненулевых строк |
Опции htseq
-f формат bam или sam
-i атрибут GFF
-s Цепь (yes, no, reverse)
-m алгоритм выбора того, относятся ли прочтения к гену (union, intersection-strict, intersection-nonempty])
ENSG00000072274.8 20495
ENSG00000252174.1 4
__no_feature 478
__not_aligned 154
__alignment_not_unique 42
Большинство чтений легли в область гена ENSG00000072274.8 – это трансферриновый рецептор, он нужен для транспорта трансферрина в комплексе железа через мембрану. 478 генов не попали в границы какого-либо гена. Это может быть в связи с тем, что риды содержат неспецифичную последовательнотсь (поли A хвост, другие повторы).
©Бакулин Артемий, 2018