import prody
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
prot = prody.parsePDB("7AOT")
@> Connecting wwPDB FTP server RCSB PDB (USA). @> 7aot downloaded (7aot.pdb.gz) @> PDB download via FTP completed (1 downloaded, 0 failed). @> 2247 atoms and 1 coordinate set(s) were parsed in 0.05s.
betas = []
names = []
for residue in prot.iterResidues():
if "CA" in residue.getNames():
mean_beta = np.mean(residue.getBetas())
betas.append(mean_beta)
names.append(residue)
betas = np.array(betas)
names = np.array(names)
<ipython-input-3-db3fa53bc7a3>:9: VisibleDeprecationWarning: Creating an ndarray from ragged nested sequences (which is a list-or-tuple of lists-or-tuples-or ndarrays with different lengths or shapes) is deprecated. If you meant to do this, you must specify 'dtype=object' when creating the ndarray. names = np.array(names)
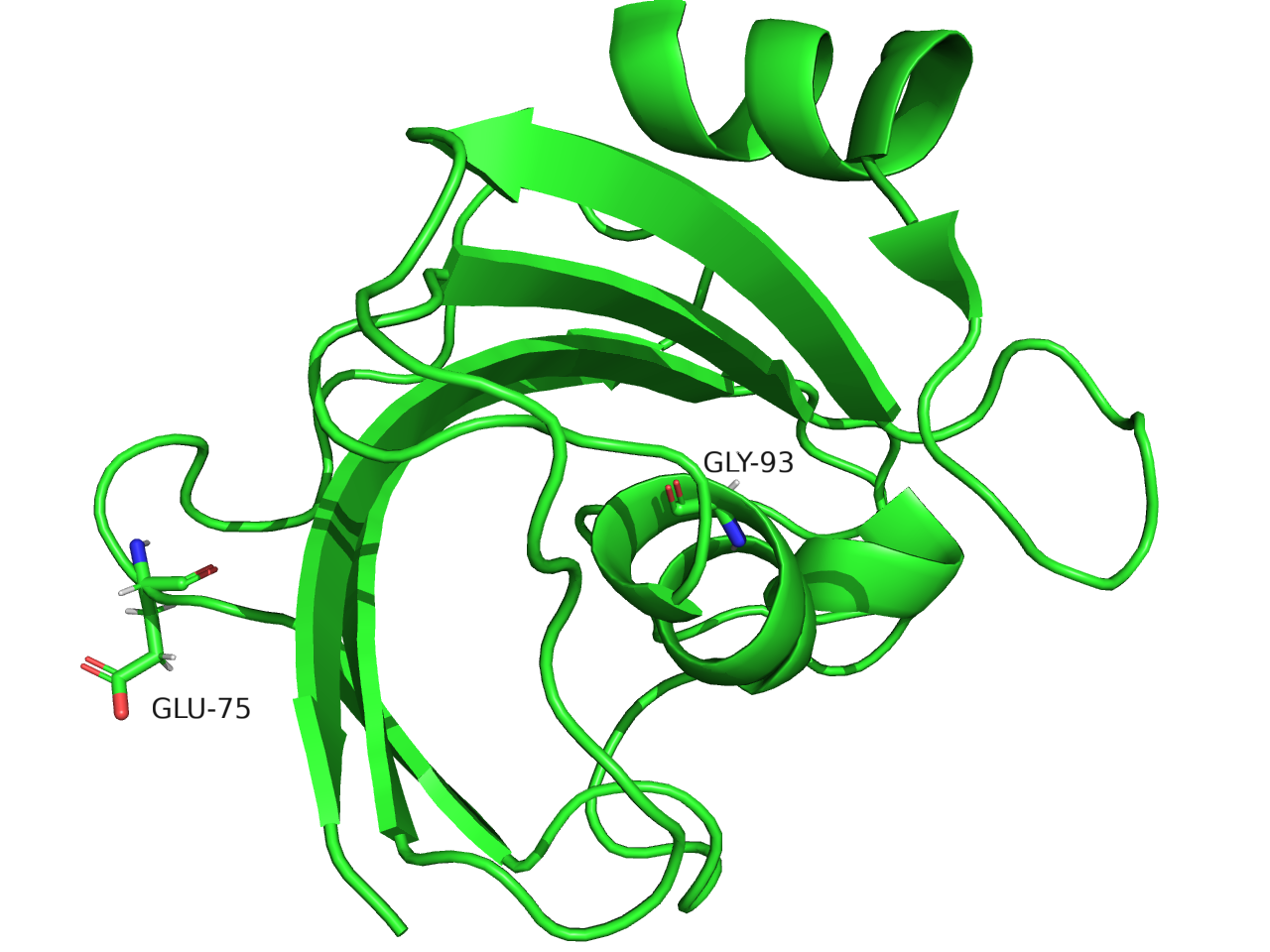
Остаток с максимальным средним b-фактором
max_res = names[np.argmax(betas)]
print(max_res)
GLU 75
Остаток с минимальным средним b-фактором
min_res = names[np.argmin(betas)]
print(min_res)
GLY 93
В белке эти остатки расположены следующим образом:
Остаток с минимальным значением фактора является частью альфа-спирали. Его боковой радикал представлен атомом водорода, то есть у этой аминокислоты у самой по себе мало степеней свободы движения, и она является частью стабильной структуры.
Остаток с максимальным значением фактора принадлежит неупорядоченному фрагменту структуры белка, а также он имеет длинный радикал экспонированный наружу. Всё это делает остаток очень подвижным.
Распределение значений фактора бета вдоль углеродного скелета молекулы выглядит следующим образом:
pd.DataFrame({'atom': max_res.getNames()[[1,4,5,6]],
'beta':max_res.getBetas()[[1,4,5,6]]})
| atom | beta | |
|---|---|---|
| 0 | CA | 30.18 |
| 1 | CB | 36.95 |
| 2 | CG | 53.87 |
| 3 | CD | 60.22 |
То есть чем дальше атом находится от альфа атома, тем выше его b-фактор.
prot_sele = prot.select("protein") # Only aminoacids
prot_center = prody.calcCenter(prot_sele, weights=prot_sele.getMasses())
betas = []
dists = []
for residue in prot.iterResidues():
if "CA" in residue.getNames():
mean_beta = np.mean(residue.getBetas())
res_center = prody.calcCenter(residue, weights=residue.getMasses())
dist = prody.calcDistance(prot_center, res_center)
betas.append(mean_beta)
dists.append(dist)
betas = np.array(betas)
dists = np.array(dists)
Зависимость значения B-фактора от расстояния до центра масс, представлена ниже:
plt.scatter(dists, betas)
plt.ylabel("B-фактор")
plt.xlabel("Расстояние до центра масс")
plt.title("Диапазон значений B-фактора от 5 до 45")
plt.ylim(5, 45)
plt.show()

from sklearn.preprocessing import PolynomialFeatures
from sklearn.pipeline import Pipeline
from sklearn.linear_model import LinearRegression
polynomial_regression = Pipeline([
("poly_features", PolynomialFeatures(degree=2, include_bias=False)),
("lin_reg", LinearRegression())
])
B_model = polynomial_regression.fit(dists[betas<15].reshape(-1, 1), betas[betas<15].reshape(-1, 1))
dists_mesh = np.arange(0, 22, 0.01).reshape(-1, 1)
betas_predicted = B_model.predict(dists_mesh)
plt.scatter(dists[betas<15], betas[betas<15])
plt.ylabel("B-фактор")
plt.xlabel("Расстояние до центра масс")
plt.title("Диапазон значений B-фактора от 5 до 15")
plt.plot(dists_mesh, betas_predicted, c='r')
plt.ylim(5, 15)
plt.show()

Мы видим, что за рядом исключений, значения B-фактора монотонно зависят расстояния аминокислоты до центра масс. Более того эту зависимость можно характеризовать, как квадратичную.
Исключением из наблюдаемой зависимости является группа остатков со значениями B-фактора больше 15. Они находятся на большом расстоянии центра масс. Высокие значения B-фактора характеризуют высокую подвижность этих остатков. Возможно они принадлежат либо линкерным участкам на поверхности глобулы, либо концам аминокислотной последовательности.
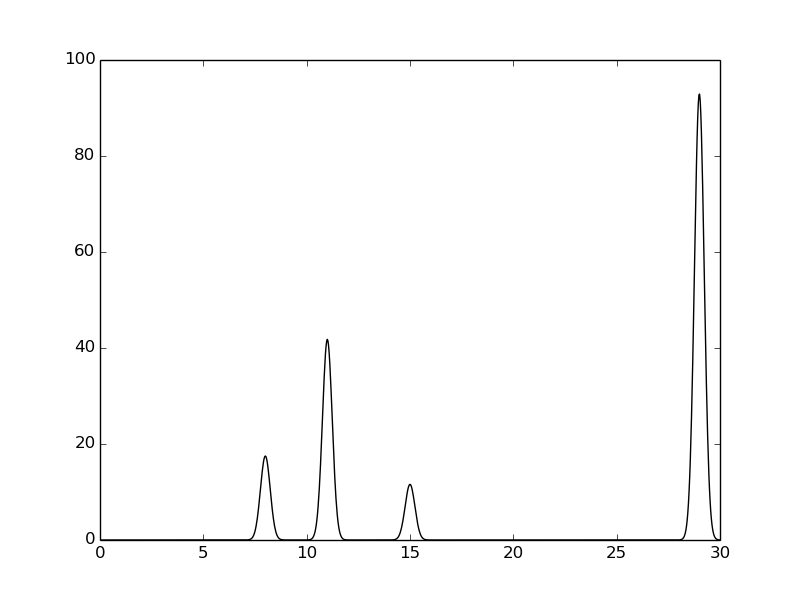
В этом задании мы провели моделирование одномерного PCA эксперимента. Для этого мы сначала задали электронную плотность и разложили её в ряд Фурье с помощью команды:
compile_func.py -g 17.5,3,8+41.8,3,11+11.6,3,15+93,3,29Затем мы проводили восстановление электронной плотности, используя разное количество гармоник.
Плохое качество – определить положение атомов невозможно
Среднее качество – положения первого и третьего атомов плохо определяются
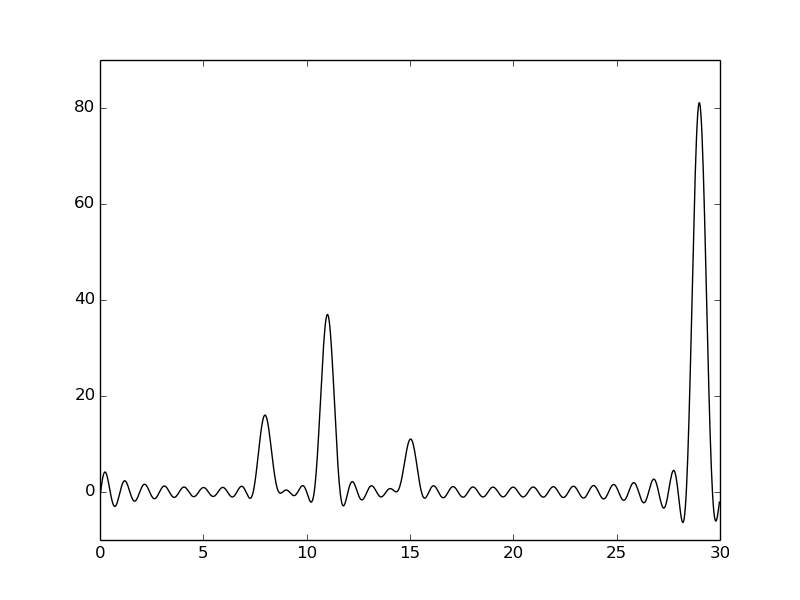
Хорошее качество – можно точно определить положение всех атомов
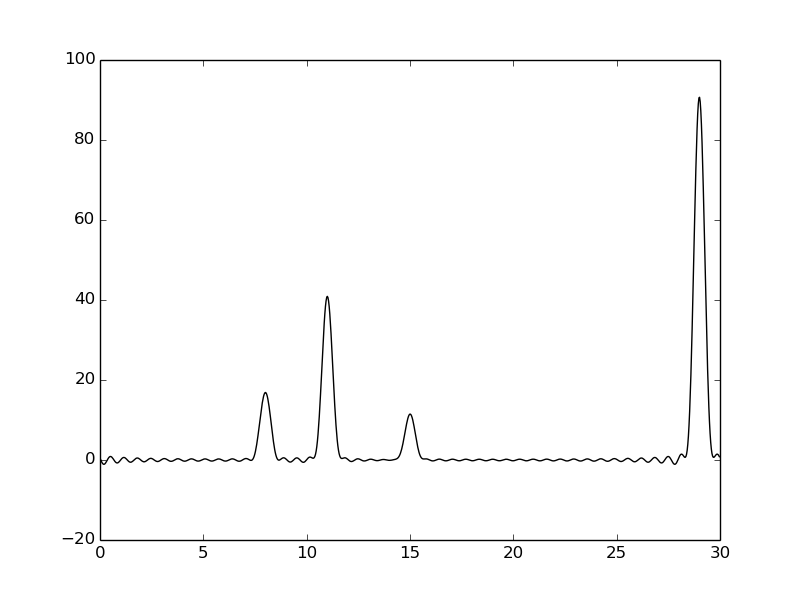
Хорошее качество – можно точно определить положение всех атомов
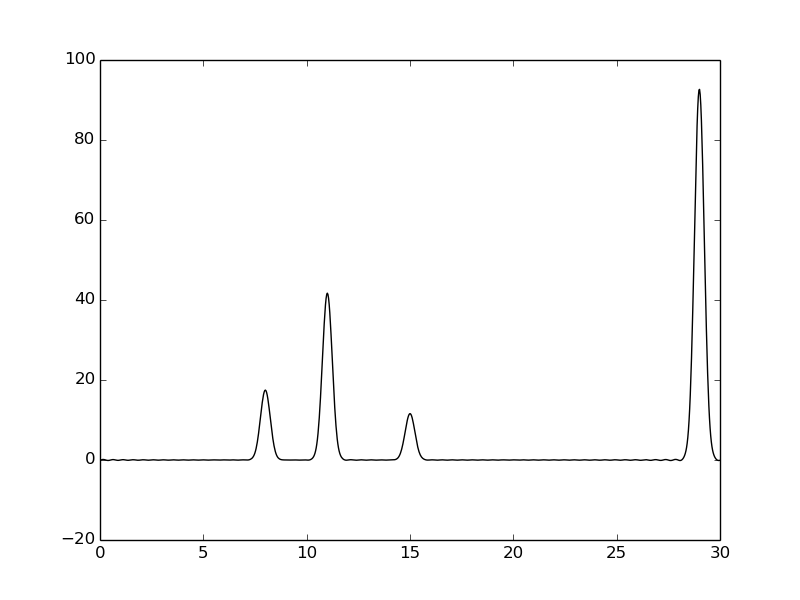
Хорошее качество – можно точно определить положение всех атомов
При увеличении количества гармоник качество сигнала возрастает, при этом сигналы становятся полностью различимы уже при 30 гармониках.
Мы также изучили, как использование неполного набора гармоник влияет на восстановление функции.
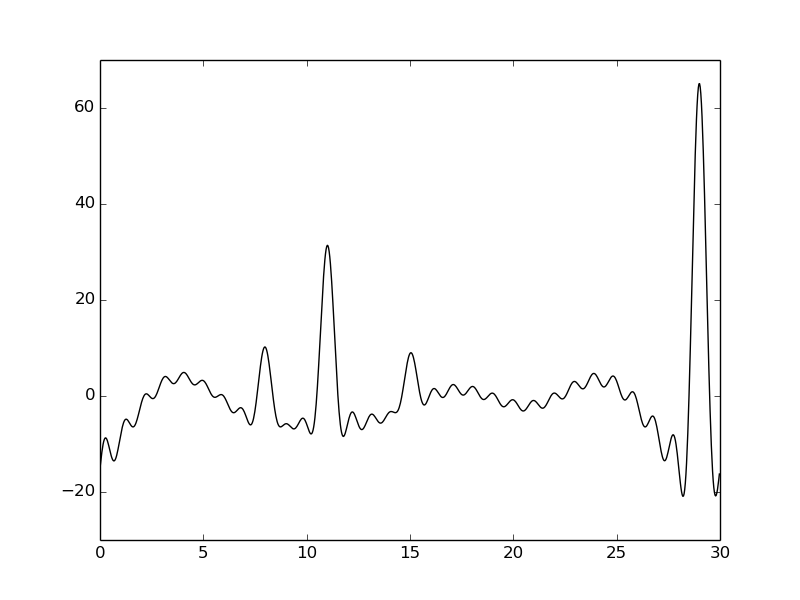
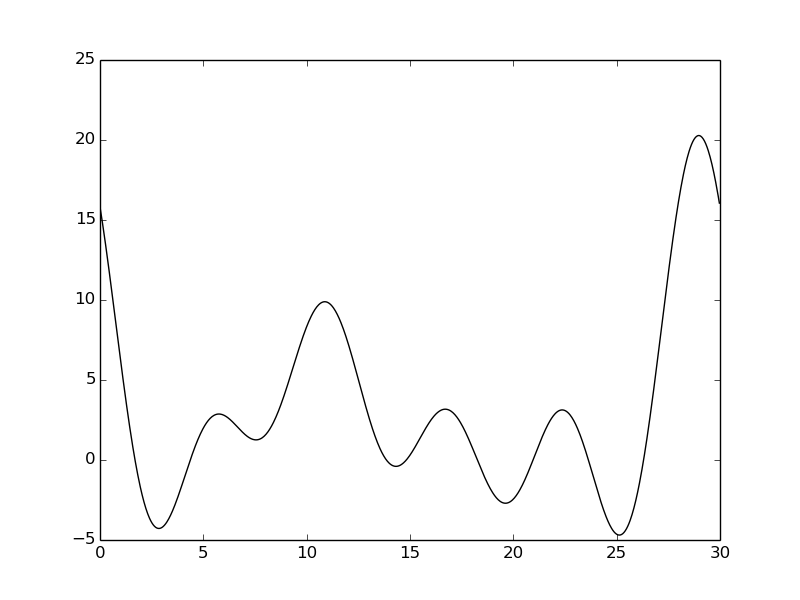
По сравнению с плотностью, построенной на отрезке 0-30, качество сильно ухудшилось. Первый и третий пики не разобрать. Наблюдается сильная осцилляция фона, который в норме должен быть равен нулю.
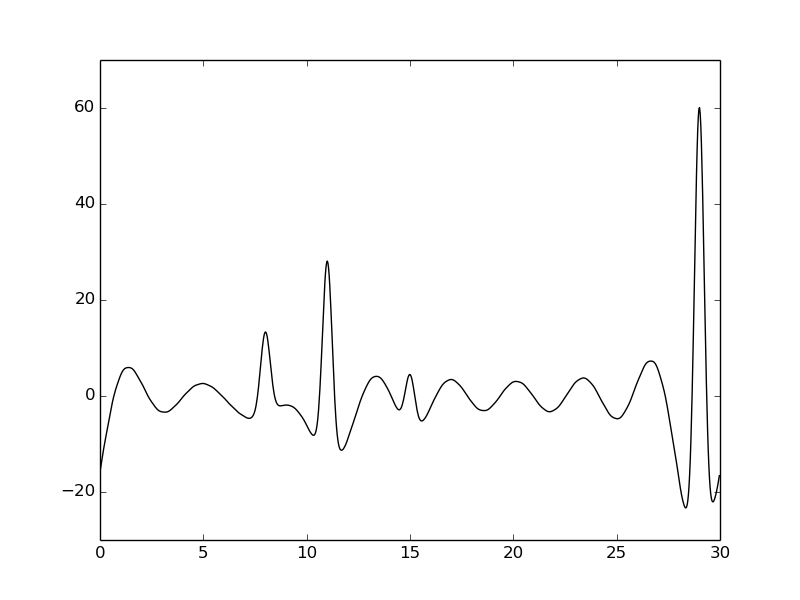
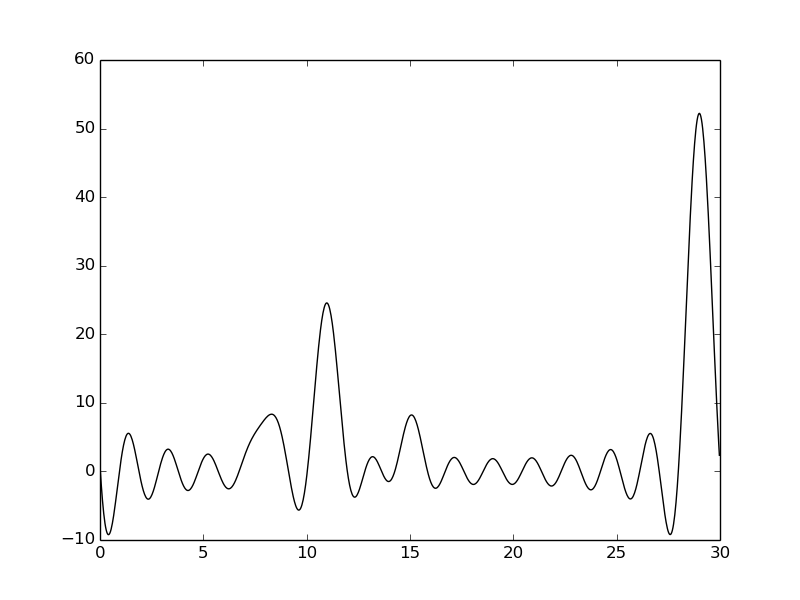
По сравнению с плотностью, построенной на отрезке 0-60, качество сильно ухудшилось. Первый и третий пики не разобрать.
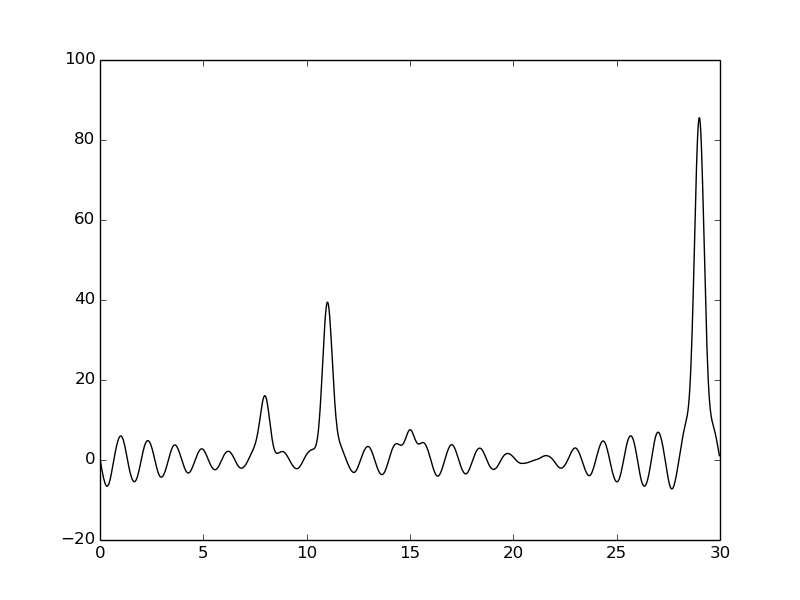
По сравнению с плотностью, построенной на отрезке 0-60, качество ухудшилось. Третий пик не разобрать.
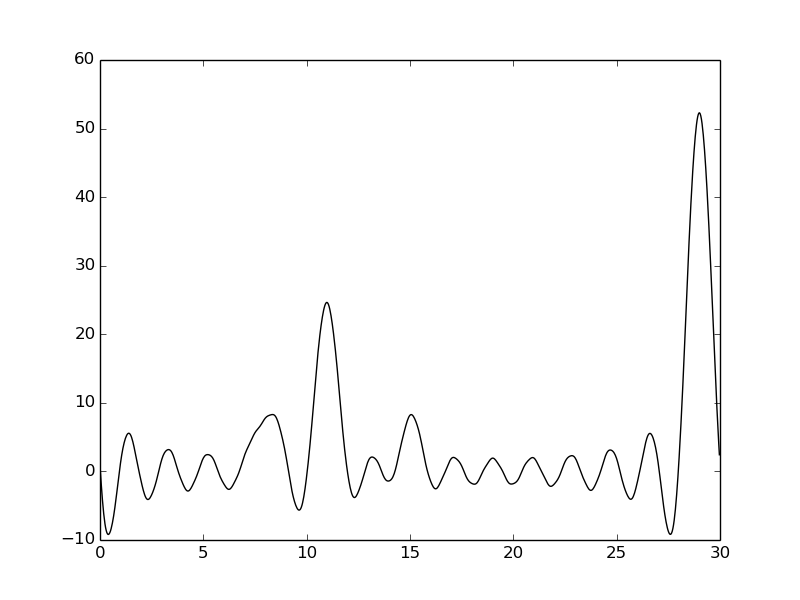
В сравнении с плотностью, построенной на отрезке 0-15, почти ничего не изменилось.
Для хорошего восстановления электронной плотности неободим весь набор гармоник на отрезке. Если пропустить всего лишь 5 гармоник в начале или в середине – качество сигнала становится средним или плохим.
Мы также исследовали, как добавление шума влияет на восстановление электронной плотности
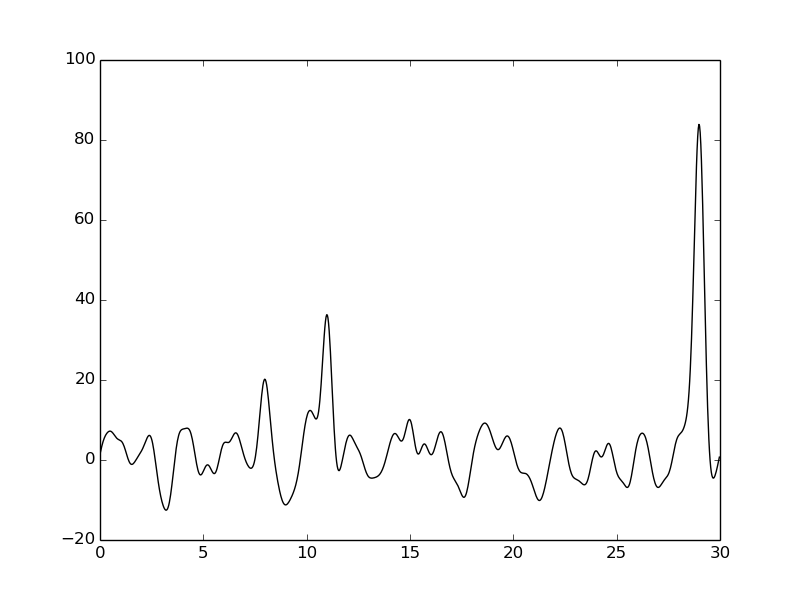
Качество очень сильно ухудшается. Первые три пика почти не различимы.
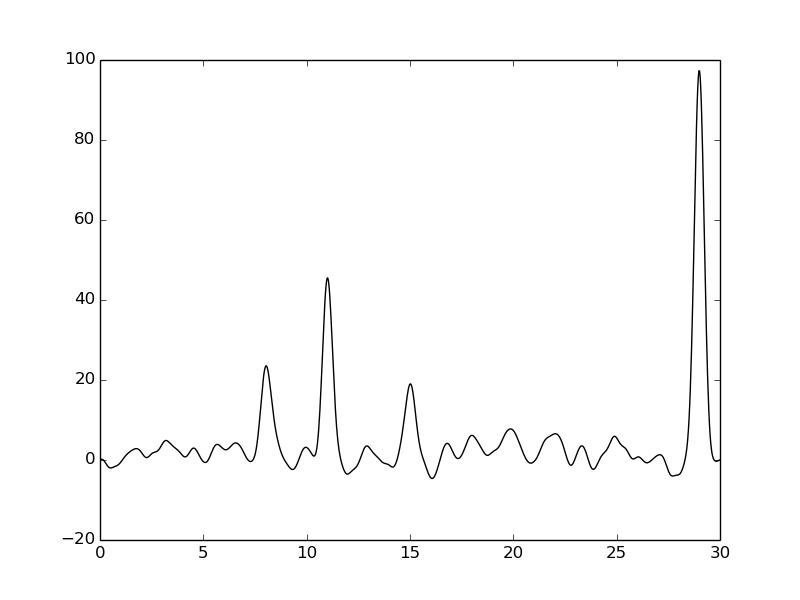
Качество не так сильно ухудшается,в целом остается хорошим. Все пики различимы.
Добавление шума по фазам сильнее влияет на сигнал, чем добавление шума по амплитудам. Даже при добавлении значительного количества шума по амплитудам качество остается хорошим.