Практикум 13. Алгоритмы и программы множественного выравнивания.
Сравнение выравнивания одних и тех же последовательностей тремя разными программами Muscle, Mafft, Tcoffee
В рамках практикума проводились выравнивания последовательностей из семейства Acyl-CoA oxidase (AC Pfam: PF01756). Выравнивания осуществлялись тремя разными программами Muscle, Mafft, Tcoffee. Затем результаты сравнивались между собой.
Сравнение множественного выравнивания с помощью программы Muscle и с помощью программы Mafft
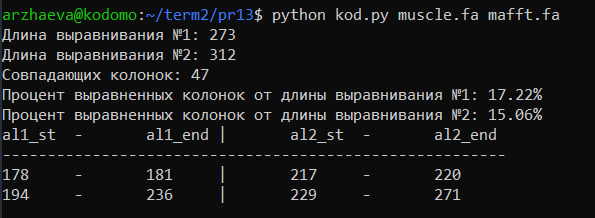
Если блок описывается так: (s1,f1)=(s2,f2), то на рисунке:
Выравнивание 1 - Muscle
Выравнивание 2 - Mafft
p.s. за код для сравнения выравниваний спасибо Михаилу Изгагину
Можно заметить, что блоки позиций, которые являются идентичными для обоих выравниваний, присутствуют. Oдинаково выровненных колонок, не входящих в блоки, нет (так как мы считаем блоком участки от 2 позиций и больше). Второй блок является достаточно большим, что говорит о локальном совпадении выравниваний.
Сравнение множественного выравнивания с помощью программы Muscle и с помощью программы T-Coffee

Если блок описывается так: (s1,f1)=(s2,f2), то на рисунке:
Выравнивание 1 - Muscle
Выравнивание 2 - T-Coffee
p.s. за код для сравнения выравниваний спасибо Михаилу Изгагину
Oдинаково выровненных колонок, не входящих в блоки, нет. Можно заметить, что блоки позиций, которые являются идентичными для обоих выравниваний, также присутствуют. Здесь их больше, а также длина блоков увеличилась.
Это говорит о том, что сходство алгоритмов выравнивания с помощью программ Muscle и T-Coffee больше, чем с помощью программ Muscle и Mafft.
Проект Jalview с 3 вышеописанными выравниваниями и последовательностями, которые выравнивались (то есть исходные):
Построение выравнивания по совмещению структур и сравните его с выравниванием программой MSA
Для выравнивания были выбраны:
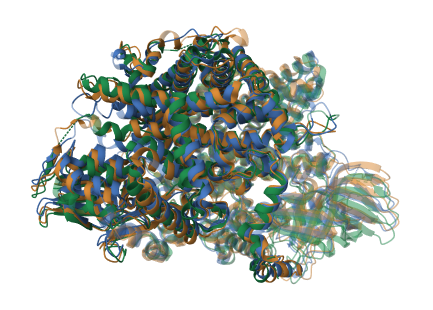
На рисунке 3 можно изображение совмещения трёх структур:
Также с помощью программы Muscle было проведено множественное выравнивание. Результаты сравнения двух выравниваний представлены на рис. 4:

Если блок описывается так: (s1,f1)=(s2,f2), то на рисунке:
Выравнивание 1 - Muscle
Выравнивание 2 - выравнивание по совмещению структур
p.s. за код для сравнения выравниваний спасибо Михаилу Изгагину
Из рис.4 видно, что присутствуют блоки одинаково выровненных колонок. Есть одинаково выровненные колонки, не входящие в блоки: позиция 683 в выравнивании 1 и позиция 677 в выравнивании 2.
Можно сделать вывод, что данные способы выравнивания могут быть использованы. Также можно сказать, что белки из одного семейства доменов обладают совпадающим участком пространственной структуры, что скорее всего коррелирует с выполняемой ими функцией. С помощью совмещения пространственных структур, а также сайта PDB можно узнать, что одинаково выровненные блоки соответсвуют как альфа спирали (блок с колонками 13-86), так и бета листу (блок с колонками 107-203 для выравнивания 1 и 109-205 для выравнивания2)
Проект Jalview с двумя вышеописанными выравниваниями:
Описание одной из программ MSA
Описание MUSCLE
MUSCLE (Multiple Sequence Comparison by Log-Expectation) - компьютерая программа для создания множественных выравниваний последовательностей белков, выпущенная в 2004 году.
Наиболее естественной формулировкой вычислительной задачи множественного выравнивания является определение модели эволюции последовательности, путём нахождения наиболее вероятного направленного графа, в котором ребра представляют собой редактирования, а конечные узлы — наблюдаемые последовательности. Но это всё-таки далеко от реальной жизни. Обычно используют поиск множественного выравнивания, которое оптимизирует сумму оценок пар.
Алгоритм осуществляется в 3 этапа:
- Этап 1 - произвести множественное выравнивание, делая упор на скорость, а не на точность
Расстояние k -мера вычисляется для каждой пары входных последовательностей, давая матрицу расстояний D1, на основе которой с помощью UPGMA (невзвешенный средний), образуется бинарное дерево TREE1. Прогрессивное выравнивание MSA1 строится путем следования порядку ветвления TREE1.
- Этап 2 - MUSCLE переоценивает дерево с использованием расстояния Кимуры, которое точнее, но требует выравнивания, так как 1 этап приблизительный и может привести к неоптимальному дереву.
Расстояние Кимуры для каждой пары входных последовательностей вычисляется из MSA1, давая матрицу расстояний D2, которая кластеризуется с помощью UPGMA, образуя бинарное дерево TREE2. Прогрессивное выравнивание на основе TREE2 создаёт множественное выравнивание MSA2.
- Этап 3 - уточнение и корректировка неточностей
Выбирается ребро из TREE2. TREE2 делится на два поддерева путем удаления ребра. Выравнивается каждое поддерево отдельно и оценивается улучшение. Если оно есть новое выравнивание сохраняется, в противном случае оно отбрасывается.
Источник: Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004 Mar 19;32(5):1792-7. doi: 10.1093/nar/gkh340. PMID: 15034147; PMCID: PMC390337.
p.s. за код для сравнения выравниваний спасибо Михаилу Изгагину