Практикум 14. Сборка de novo
Скачивание одноконцевых чтений
Код доступа проекта по секвенированию бактерии Buchnera aphidicola str. Tuc7: SRR4240361
Чтения получены в данном проекте по технологии Illumina
Код для скачивания чтений в рабочую директорию (pr14):
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/001/SRR4240361/SRR4240361.fastq.gz
Подготовка чтений программой trimmomatic
Для начала необходимо сохранить адаптеры для Illumina в один файл с помощью данной команды:
cat ../../adapters/* > adapters.fasta
Затем, с помощью trimmomatic были удалены адаптеры (2:7:7 - параметры, задающие соответственно максимальное количество мисметчей в последовательности адаптера, порог качества для обнаружения палиндромных адаптеров (2 цепях), порог качества для обнаружения просто адаптера (на одной цепи), не являющегося палиндромом):
TrimmomaticSE SRR4240361.fastq.gz trim.fq.gz ILLUMINACLIP:adapters.fasta:2:7:7
Input Reads: 7272621 Surviving: 7238089 (99.53%) Dropped: 34532 (0.47%)
Адаптерами оказалось 0.47 % чтений.
Команда, с помощью которой можно узнать вес файла:
du -h trim.fq.gz
После этого были удалены с правых концов чтений нуклеотиды с качеством ниже 20, а также чтения с длиной меньше 32 нуклеотидов:
TrimmomaticSE trim.fq.gz result.fq.gz TRAILING:20 MINLEN:32
Input Reads: 7238089 Surviving: 6834335 (94.42%) Dropped: 403754 (5.58%)
Команда, с помощью которой можно узнать вес файла:
du -h result.fq.gz
Было удалено 403754 (5.58%) чтений. До очистки файл весил 192M, а после 178M.
Программа velveth
C помощью данной команды были подготовлены k-меры длины k=31 (максимально возможной). Использовались короткие и непарные чтения (short), формат файла fastq.gz
mkdir velvet
velveth ./velvet 31 -short -fastq.gz result.fq.gz
Получили 3 файла: Log, Roadmaps, Sequences
Программа velvetg
C помощью команды velvetg была осуществлена сборка на основе ранее полученных k-меров
velvetg velvet/ #на выходе получаем 5 файлов
Final graph has 477 nodes and n50 of 25683, max 49238, total 668902, using 0/6834335 reads
N50: 25683
Команда для нахождения трех самых длинных контигов и их покрытия:
grep '^>' contigs.fa | tr '_' ' ' | sort -k4nr | head -3
>NODE 6 length 49238 cov 26.660851
>NODE 2 length 45555 cov 26.450466
>NODE 34 length 43866 cov 23.514977
Максимальная длина у соответсвующих контигов составила: 49238, 45555, 43866
Покрытие соответсвенно составило: 26.660851, 26.450466, 23.514977
Затем были изучены контиги с аномально большим или аномально малым покрытием (более чем в 5 раз отличающимся от медианного):
grep '^>' contigs.fa | wc -l #208 контигов всего
grep '^>' contigs.fa | tr '_' ' ' | sort -k6nr > file.txt #отсортированный файл
awk 'NR == 104 || NR == 105 {sum += $6; count++} END {print sum/count}' file.txt #нахождение медианы
Медиана: 11.975
awk '$6 > 59.875 || $6 < 2.395' file.txt
>NODE 78 length 47 cov 90.744682
>NODE 91 length 33 cov 76.636360
>NODE 95 length 31 cov 64.903229
>NODE 185 length 48 cov 62.541668
>NODE 391 length 63 cov 2.238095
Таким образом, было найдено 4 контига с аномально большим покрытием и 1 с аномально малым покрытием.
Интересно заметить, что контиг с аномально низким покрытием имеет длину всего лишь 63, хотя можно было бы предположить, что с его покрытием он должен быть сильно длинее. Наоборот, контиги с максимальным покрытием имеют маленькую длину, что вполне логично (напомню, что максимальная длина составляет 49238, а у данных контигов она от 31 до 48)
Анализ
Для выполнения этого задания я выбрала Buchnera aphidicola str. APS (Acyrthosiphon pisum). Хромосома - NZ_AP036055.1
Получили последовательности контигов с максимальной длиной с помощью: (выбираем нужные строчки, а именно от строчки с названием контига и до следующего символа '>' в начале строки, выводим все, кроме последней строчки, которая соответствует уже новому контигу)
sed -n '/>NODE_2_length_45555_cov_26.450466/,/^>/{p}' contigs.fa | head -n -1
sed -n '/>NODE_6_length_49238_cov_26.660851/,/^>/{p}' contigs.fa | head -n -1
sed -n '/>NODE_34_length_43866_cov_23.514977/,/^>/{p}' contigs.fa | head -n -1
С помощью megablast были проведены выравнивания 3 самых длинных контигов, полученных ранее и хромосомы Buchnera aphidicola str. APS
Контиг NODE 2. Для него:
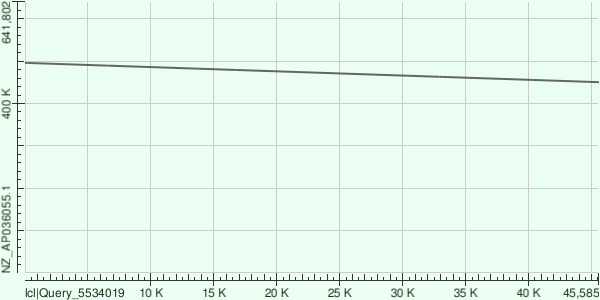
Можно предположить, что так как мы запускаем выравнивание для двух штаммов одной бактерии, то карта локального сходства должна показать точное соотвествие изучаемого рида какому-то фрагменту хромосомы взятой бактерии. На рисунке 1 можно заметить, что карта локального сходства полностью подтверждает то, что в хромосоме выбранной бактерии содержится участок идентичный исследуемому контиг, так как видно непрерывную ровную линию, которая говорит о взаимном соответствии.
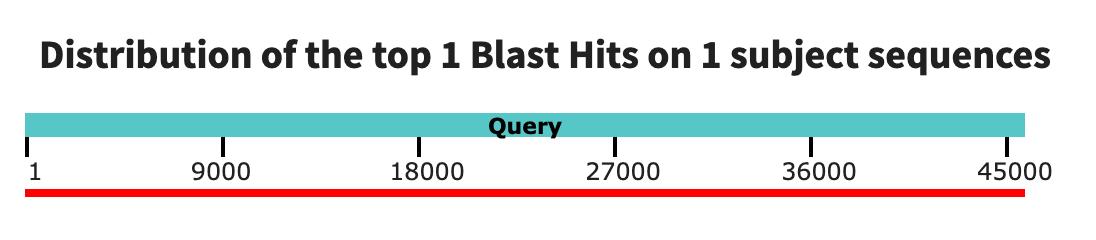
На рисунке 2 можно заметить, что контиг практически полностью (99%) ложится на взятую хромосому.
Контиг NODE 6. Для него:
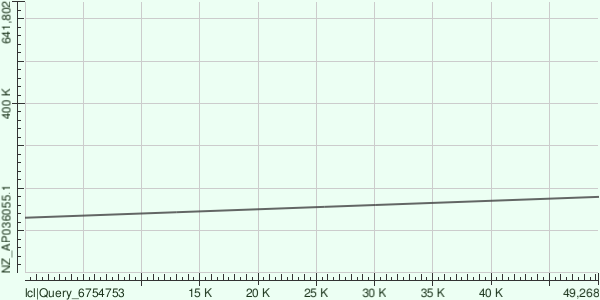
На рисунке 3 аналогично описанному выше можно заметить, что карта локального сходства также полностью подтверждает то, что в хромосоме выбранной бактерии содержится участок идентичный исследуемому контигу.

На рисунке 4 можно заметить, что контиг практически полностью (99%) ложится на взятую хромосому.
Контиг NODE 34. Для него:
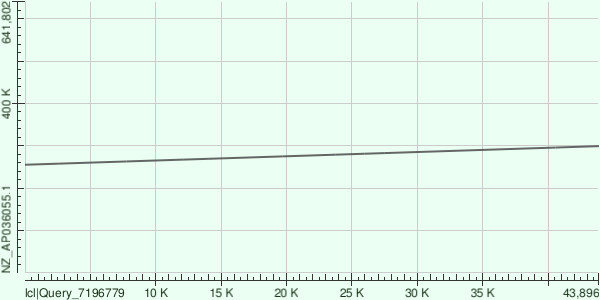
На рисунке 5 аналогично можно заметить, что карта локального сходства также полностью подтверждает то, что в хромосоме выбранной бактерии содержится участок идентичный исследуемому контигу.
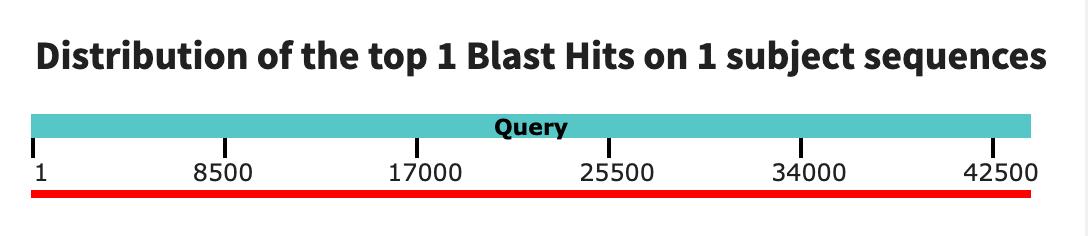
На рисунке 6 можно заметить, что контиг практически всей своей длинной (99%) ложится на взятую хромосому.