Практикум 2. Филогенетическая реконструкция и сравнение деревьев
В первую очередь было создано выравнивание последовательностей цитохромов B у животных из Практикума 1. Я пользовалось kodomo, поскольку на kodomo к EMBOSS подключён банк Swiss-Prot под именем sw
Код для создания выравнивания:
seqret @cyb.list cyb.fasta #cyb.list - файл с идентификаторами в Swiss-Prot (sw:cyb_human)
muscle -align cyb.fasta -output cyb-alignment.fasta #используем fasta файл и программу muscle для выравнивания
Все деревья были визуализированы с помощью iTOL
FastME
С помощью скрипта выравнивание было переведено в формат "phylip-relaxed", на выходе был получен файл cyb.phy, который использовался далее
Реконструкция дерева на kodomo программой FastME (алгоритм которой основан на принципе минимальной эволюции (Minimum evolution)):
fastme -i cyb.phy -o pd.tre -pp #модель p-distance (оценивает: число отличий/длину = доля несовпадающих позиций)
fastme -i cyb.phy -o mtrev.tre -pM #модель MtREV (матрица с относительными скоростями замен аминокислоты одной на другую)
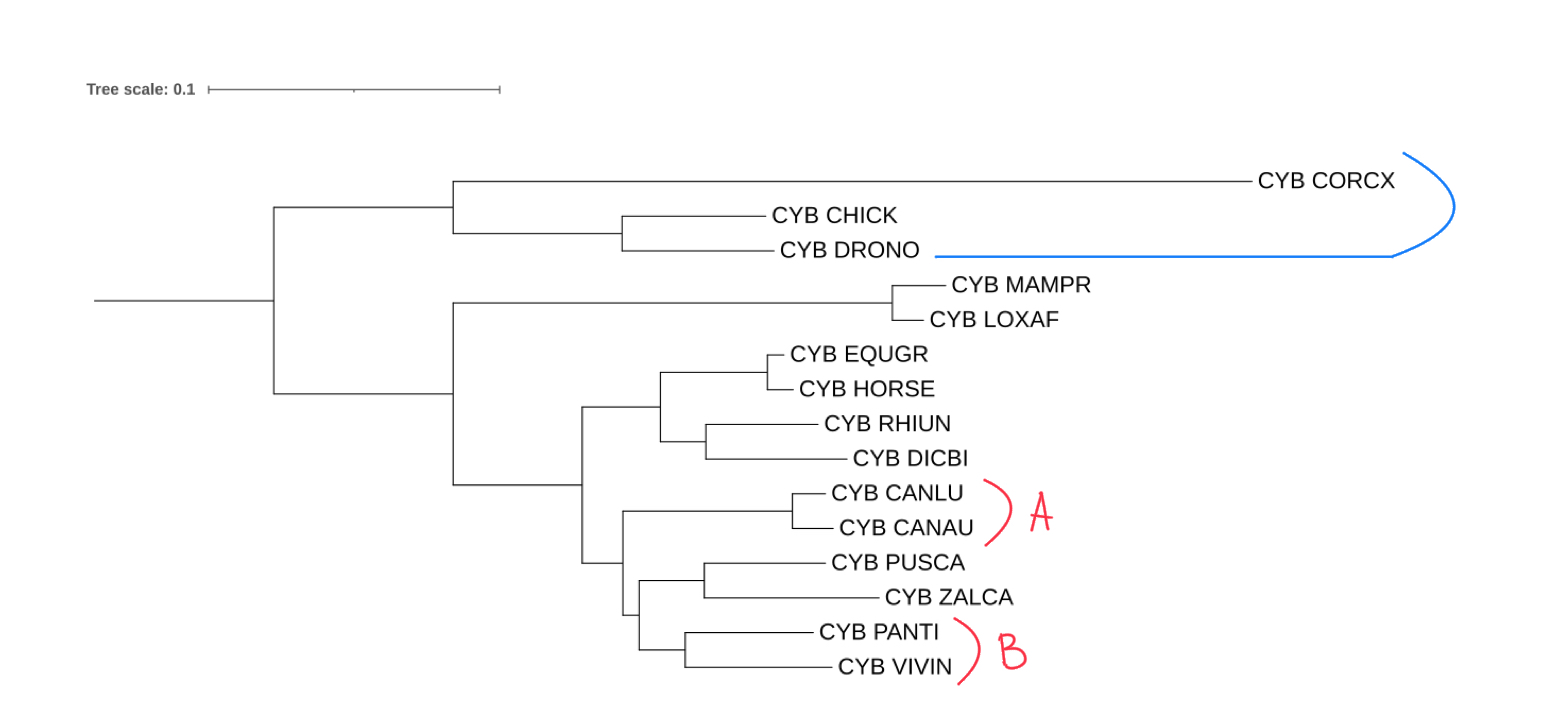
На рисунке 1 представлено филогенетическое дерево, которое имеет ряд отличий от исходного, построенного в рамках Практикума 1:
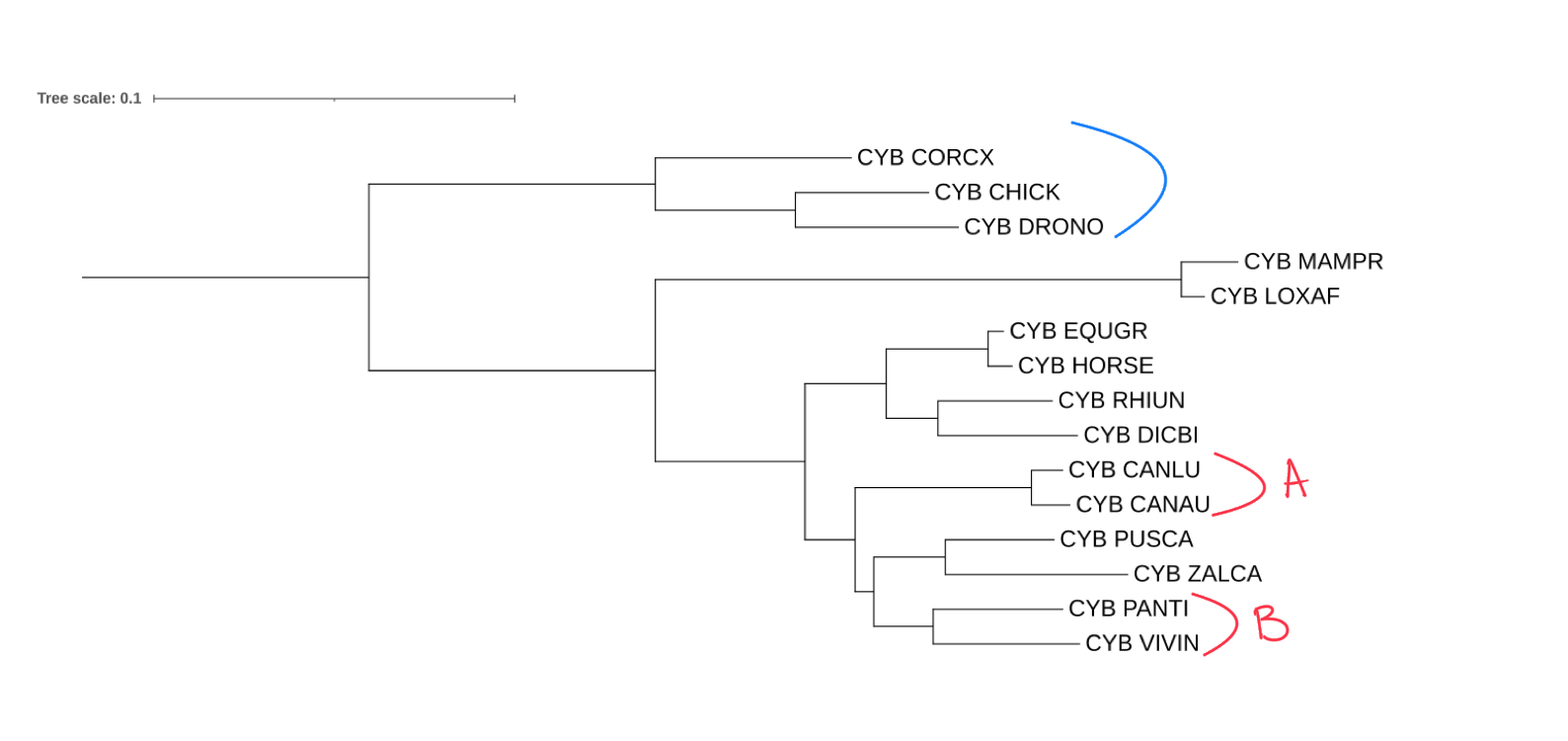
На рисунке 2 представлено филогенетическое дерево, которое имеет ряд отличий от исходного, построенного в рамках Практикума 1, но очень похоже на рисунок 2:
IQ-Tree
Реконструкция дерева на kodomo программой IQ-Tree (алгоритм реконструкции - максимальное правдоподобие (Maximum Likelihood)), которая с помощью ModelFinder автоматически выбирает эволюционную модель (файл cyb.phy.iqtree), в данном случае использовалась mtMAM+G4 (модель, представляющая собой матрицу скоростей замен аминокислот митохондрий позвоночных и модель, учитывающая гетерогенность скоростей мутаций в разных позициях):
iqtree -s cyb.phy
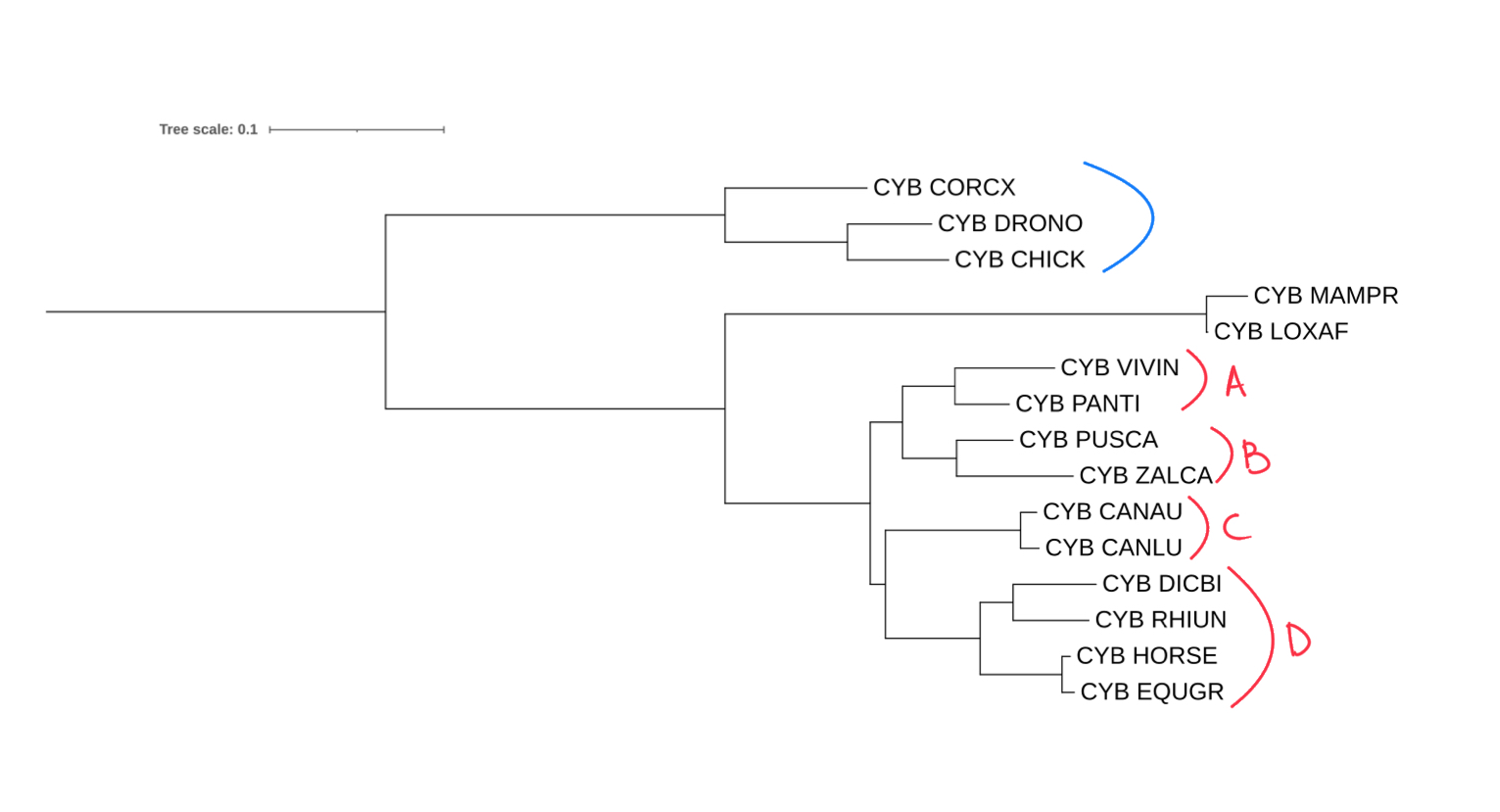
На рисунке 3 представлено филогенетическое дерево, которое имеет ряд отличий от исходного, построенного в рамках Практикума 1: