Практикум 8. Сигналы и мотивы
1. Описание мотива в белках паттерном
Для выполнения задания была выбрана энолаза (Phosphopyruvate hydratase). Мнемоника Swiss-Prot: ENO. Данный фермент катализирует превращение 2-фосфоглицерата в фосфоенолпируват в ходе гликолиза. Принадлежит к семейству лиаз, а именно к гидролиазам, которые расщепляют связи углерод-кислород.
Для выравнивания использовались следующие последовательности белков: ENO_ECOLI, ENO_BURMA, ENO_NEIMA, ENO_PASMU, ENO_ROSDO, ENO_SHEDO, ENO_SERP5, ENO_PARDP
Количество белков с такой мнемоникой в файле bacteria-sw.fasta: 748
Мнемоники бактерий:
Код для создания выравнивания:
seqret @eno.list eno.fasta #eno.list - файл с идентификаторами в Swiss-Prot (sw:eno_ecoli)
muscle -align eno.fasta -output eno-alignment.fasta #используем fasta файл и программу muscle для выравнивания
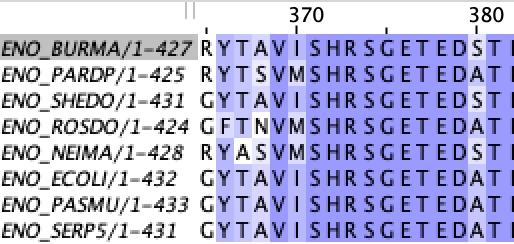
Выравнивание представлено здесь.
Для составления паттерна был выбран консервативный участок без гэпов: позиции 369-379
Паттерн: V-[IM]-S-H-R-S-G-E-T-E-D
Поиск программой fuzzpro по этому паттерну среди всех белков бактерий из Swiss-Prot:
fuzzpro /P/y24/term4/bacteria-sw.fasta -pattern 'V-[IM]-S-H-R-S-G-E-T-E-D' eno.fuzzpro
- Общее число находок: 391
- Число находок с выбранной мнемоникой (ENO): 385
- Число ложноположительных результатов: 6
- Число ложноотрицательных результатов (ненайденные белки с выбранной мнемоникой): 748 - 385 = 363
Поскольку число ненайденных белков оказалась большим, для усиления паттерна я решила расширить его, а также попробовать найти другой консервативный участок, но, к сожалению, 391 - максимальное число находок, которого мне удалось достичь.
2. Поиск мотивов в белках программой MEME и поиск этих мотивов в банке
Код для запуска программы MEME:
meme eno.fasta -protein -mod oops -nmotifs 3 -minw 8 -maxw 15 -o meme_output
Расшифровка опций и параметров запуска:
Результат MEME представлен здесь.
В результате анализа набора из 8 последовательностей энолаз (ENO) с помощью программы MEME было обнаружено 3 консервативных мотива в каждом белке. Мотивы имеют очень низкое E-value (в диапазоне: 10⁻⁶⁰ — 10⁻⁶⁷), что свидетельствует о высокой статистической значимости находок (вероятность случайного обнаружения подобных мотивов в случайных последовательностях очень низкая). Для каждой аминокислотной последовательности p-value в диапазоне: 10⁻⁴⁴ - 10⁻⁴⁵, что подтверждает высокую достоверность локализации мотивов в конкретных позициях этого белка.
Код для запуска программы MAST:
mast meme_output/meme.html /P/y24/term4/bacteria-sw.fasta -o mast_output
Результат MAST представлен здесь.
Число находок с E-value<10 составило 775. Порог позиционного p-value составил 0.0001. Все находки имеют низкое E-value, что подтверждает их статистическую значимость.
- Общее число находок: 775
- Число находок с выбранной мнемоникой (ENO): 747
- Число ложноположительных результатов: 775 - 747 = 28
- Число ложноотрицательных результатов (ненайденные белки с выбранной мнемоникой): 748 - 747 = 1
Таким образом, не нашелся всего 1 белок. Этот результат гораздно лучше, чем при ручном составлении мотива по выравниванию.
3. Поиск последовательности Шайна — Дальгарно в геноме своего прокариота
В рамках данного задания использовался геном бактерии Leptospira borgpetersenii serovar Ceylonica
Последовательность Шайна - Дальгарно - это консервативный участок на мРНК прокариот (AGGAGG), находящийся перед старт-кодом (на расстоянии около 10-ти нуклеотидов). Он необходим для комплементарного взаимодействия с 16s рРНК, и, таким образом, обеспечивает правильное связывание мРНК с рибосомой.
Код для запуска программы fuzznuc:
fuzznuc ~/term1/genome/GCF_003516145.1_ASM351614v1_genomic.fna -pattern 'A-G-G-A-G-G' -complement -outfile SD_seq.outfile # complement - позволяет искать и на прямой, и на комплементарной цепи
Программа fuzznuc обнаружила 1243 участка (N_obs), соответствующих паттерну A-G-G-A-G-G (с учётом комплементарной цепи) в геноме бактерии.
Для того, чтобы найти ожидаемое число находок (N_exp) с учетом комплементарной цепи (умножение на 2) нужно: P(AGGAGG) * Length(genome) * 2
P(AGGAGG) = P(A)**2 × P(G)**4
Для нахождения этой вероятности, найдем сколько раз встречаются A и G в геноме, а потом вычислим вероятность.
- A: 1 187 046
- G: 807 569
- Length (genome): 3 670 231 bp
- P(A): 0,323
- P(G): 0,220
Получаем ожидаемое число находок (N_exp): 897
Чтобы оценить достоверность отличия наблюдаемого числа находок от ожидаемого воспользуемся формулой:
√(Nexp)
Рассчитанное значение статистики составило 11.55. Поскольку это значение превышает критическое 1.96 для уровня значимости α=0.05, нулевая гипотеза об отсутствии различий между наблюдаемым и ожидаемым числом находок отвергается. Следовательно, наблюдаемое число участков с паттерном AGGAGG статистически значимо отличается от случайно ожидаемого.
С помощью геномной таблицы (GCF_003516145.1_ASM351614v1_feature_table.txt) и скрипта был найден процент находок, которые располагаются в правильной позиции относительно старт-кодона.
Код для запуска скрипта:
python pr8.py SD_seq.outfile ~/term1/genome/GCF_003516145.1_ASM351614v1_feature_table.txt
Было найдено 43 находки в правильном положении из 1243. Это составляет 3.5 %. Скорее всего такой низкий полученный процент можно объяснить тем, что у некоторых организмов эта последовательность может быть немного иной, а также тем, что вероятность в геноме встретить последовательность AGGAGG достаточно велика, поэтому получилось много случайных находок.