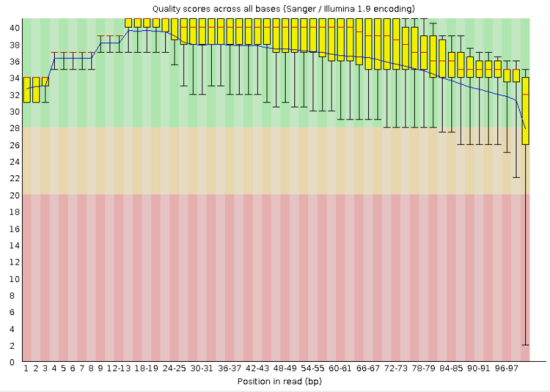
Рис.1 Изображение качества оснований из результатов FastQC до обработки Trimmomatic.
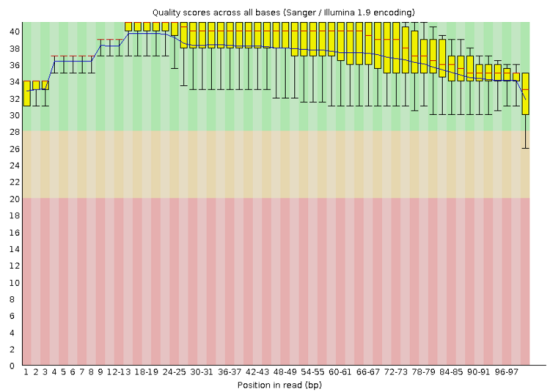
Рис.2 Изображение качества оснований из результатов FastQC после обработки Trimmomatic.
|
Меню
На главную Третий семестр |
Подготовка чтенийПеред поиском полиморфизмов проверила качество прочтений([1],Табл.3.). Оно оказалось ниже требуемого(20), поэтому надо было удалить основания с плохим качеством. Короткие последовательности(<50 пар оснований) тоже не нужны и будут удалены. ([2], Табл.3.) На рисунке 2 представлены чтения после обработки. |
| |
|
|
| Рис.1 Изображение качества оснований из результатов FastQC до обработки Trimmomatic. |
Рис.2 Изображение качества оснований из результатов FastQC после обработки Trimmomatic. |
Из рисунков 1-2 видно, что после использования Trimmomatic улучшилось качество чтений(особенно у концевых оснований). Также изменилось количество последовательностей: было 10701, стало 10534.
Картирование чтенийПолучено выравнивание прочтений и проиндексированной([3],Табл.3) рефересной последовательности с помощью алгоритма mem([4], Табл.3). Далее выравнивание было переведено в двоичный формат([5], Табл.3), отсортировано по началу в референсе([6], Табл.3) и заново проиндексировано, но уже с помощью samtools.([7], Табл.3) На геном откартировались все чтения, кроме одного(10333 чтения из 10334).([8], Табл.3)Анализ SNPБыл получен файл с полиморфизмами([9], Табл.3) и файла со списком отличий между референсом и чтениями.([10], Табл.3) В нем найдено 102 полимофизма и 6 иделей. В таблице 1 приведены три полиморфизма: один с самым большим качеством, второй с самой большой глубиной чтений и индель. В целом в файле покрытие от 1 до 98. Много(37) полиморфизмов с покрытием 1. В среднем покрытие около 13. Качество от 3,54577 до 226.13. Качество ридов было улучшено до 20. Но в итоге качество некоторых полиморфизмов заметно меньше 20. Так же у этих полиморфизмов глубина всего 1. Т.е. такой полиморфизм встретился только в одном прочтении.Табл.1. Примеры полиморфизмов.
Аннотация SNPУдалила индели из файла с полиморфизмами. Скопировала скрипты annotate_variation.pl и convert2annovar.pl в свою директорию и воспользовалась последним, чтобы получить файл для annovar.([11], Табл.3)refgene - gene-based annotation ([12], Табл.3) Табл.2. Категории полиморфизмов по месту нахождения в хромосоме.
dbsnp - filter-based annotation([13], Табл.3) С помощью скрипта выяснила, какие snp имеют rs(т.е. имеют свое имя). Только 7 штук из 102 не имеют rs. 1000 genomes - filter-based annotation([14], Табл.3) 9 полиморфизмов встречаются в геномах этой 1000 людей с частотой меньше 5%. Gwas - region-based annotation([15], Табл.3) Найдено 8 полиморфизмов, которые могут быть связанны с заболеваниями: болезнь Крона, эндометриоз, малярия, венозный тромбоэмболизм, опухоль и факторы коагуляции. Clinvar - filter-based annotation([16], Табл.3) Было найдено 3 полиморфизма: два отвечают за группу крови и один неспецифичный. Ссыслка на сводную таблицу с аннотациями по всем банкам данных.
|
© Корзина Анастасия, 2015