| |
Структуры из ЯМР- и РСА-файлов были выравнены с помощью RCSB PDB Protein Comparison Tool. Что показало, что структуры не сильно отличаются(Рис.1). Далее были выбраны водородные связи: в петле(Рис.2) и в глобуле(Рис.3).
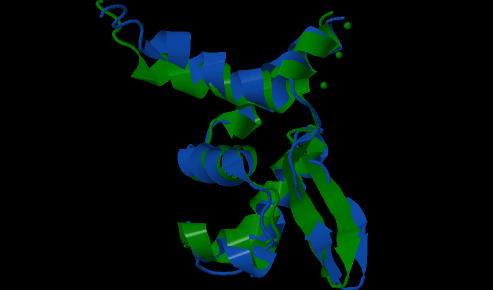 Рис.1. Выравнивание структур CzrA, полученных РСА (зеленый) и ЯМР (синий). |
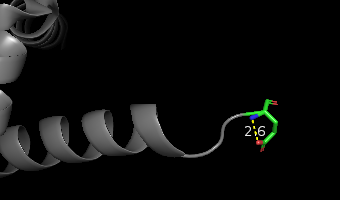 Рис.2. Водородная связь в петле, выступающей над поверхностью глобулы. |
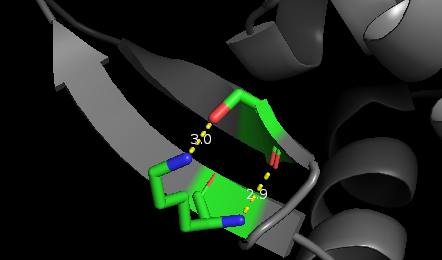 Рис.3. Водородные связи в глобуле в между остовными атомами(2.9Å) и между атомами из боковых цепей(3.0Å) . |
В таблице 1 приведены длины водородных связей из РСА- и ЯМР-файлов. Нумерация аминокислотных остатков в файлах совпадает.
Табл.1. Сравнение длин водородных связей в ЯМР- и РСА-файлах.
| Водородная связь |
Расстояние в РСА, Å |
Наличие в моделях ЯМР, % |
Минимальное расстояние в ЯМР, Å |
Максимальное расстояние в ЯМР, Å |
Среднее расстояние в ЯМР, Å |
| 103 GLU OE2 - N GLU 103 (петля) |
2.6 |
9.5 |
2.9 |
3.0 |
2.95 |
| 81 SER O - N LYS 70 (остов) |
2.9 |
9.5 |
3.4 |
3.4 |
3.4 |
| 81 SER OG - NZ LYS 70 (боковые цепи) |
3.0 |
0 |
- |
- |
- |
В ЯМР-файле ни одна выбранная водородная связь не встречается в каждой модели. При этом связь между атомами из боковых цепей не встречается ни в одной модели из ЯМР-файла. Это можно объяснить тем, что различия между РСА- и ЯМР-файлами есть уже на уровне вторичной структуры. В РСА-файле остатки SER 81 и LYS 70 входят в бета-лист, тогда как во всех моделях из ЯМР-файла они составляют петлю (Рис.1).
|