|
В данном задании необходимо определить таксономию и функцию прочтенной нуклеотидной последовательности (из практ. 6).
В качестве нуклеотидной последовательности использовалась объединенная из прямой и комплементарной обратной цепей последовательность.
Ссылка на файл с последовательностью в формате fasta. База данных: Nucleotide collection (nr/nt); Алгоритм: Megablast (highly similar sequences); Был проведен поиск, ограниченный 100-ми находками, большинство которых соответствует гену 1-ой субъединицы цитохром с-оксидазы. Поэтому можно предположить, что прочтенная последовательность имеет сходные функции, что и данный ген. |
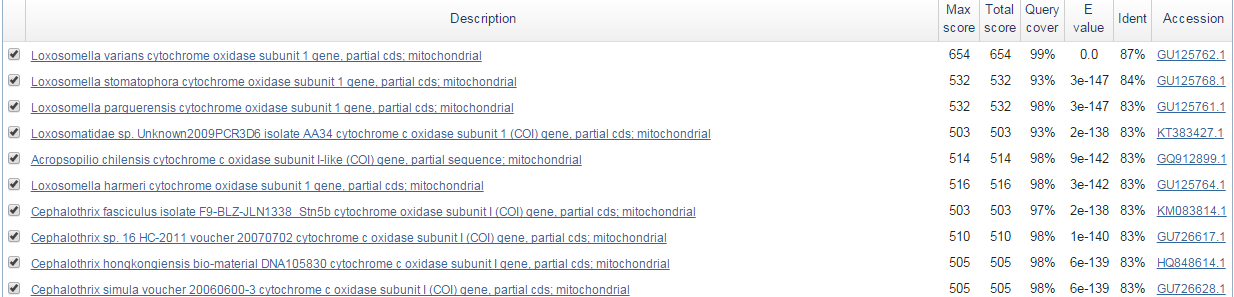
Рис.1 - Первые 10 находок алгоритма megablast
| |
|
Для построения выравнивания были взяты первые 10 находок по критерию Ident.
На рис. 2 приведено выравнивание.
Можно увидеть, что данная последовательность "вписывается" по числу замен, то есть можно назвать ее гомологом остальных.
Если посмотреть по таксономии - большинство выбранных организмов (9 из 10) относятся к надтипу Lophotrochozoa.
Визуально можно отметить, что последовательность Loxosomella varians наиболее схожа с данной, и она же выдает самый высокий показатель по Ident и Max score.
Таким образом, можно сделать вывод, что данная последовательность относится к данному виду.
Предпологаемая таксономия: Eukaryota, Protostomia, Lophotrochozoa, Loxosomatidae, Loxosomella. |
Рис. 2 - Выравнивание находок и последовательности (при нажатии картинка увеличивается)
 |
|
В данном задании необходимо было продемонстрировать сравнение трех алгоритмов blast.
В качестве последовательности была взята плазмида pNGTCDC08107 Neisseria gonorrhoeae TCDC-NG08107.
Ссылка на файл с последовательностью в формате fasta. |
Таблица 1. Параметры запуска BLAST
| Algorithm | Database | Organism | Max target sequences | Expect threshold | Word size | Max matches in a query range | Match/ Mismatch Scores | Gap Costs | Filter | Mask |
| Megablast (highly similar sequences) | Nucleotide collection (nr/nt) | Neisseria (taxid:482) - Exclude,
Neisseriaceae (taxid:481) |
1000 | 10 | 16 | 0 | 1, -2 | Linear | Low complexity regions | Mask for lookup table only |
| Discontiguous megablast (more dissimilar sequences) | Nucleotide collection (nr/nt) | Neisseria (taxid:482) - Exclude,
Neisseriaceae (taxid:481) |
1000 | 10 | 11 | 0 | 2, -3 | Existence: 5 Extension: 2 | Low complexity regions | Mask for lookup table only |
| Blastn (somewhat similar sequences) | Nucleotide collection (nr/nt) | Neisseria (taxid:482) - Exclude,
Neisseriaceae (taxid:481) |
1000 | 10 | 7 | 0 | 2, -3 | Existence: 5 Extension: 2 | Low complexity regions | Mask for lookup table only |
Таблица 2. Сравнение алгоритмов BLAST
| Алгоритм | Число находок | Примеры находок одним из алгоритмов, не найденных другим | Комментарии к данному алгоритму |
| Megablast | 3 | Candidatus Snodgrassella sp. TA7_36335 genomic sequence
Score: 36.2 bits(19), Expect: 2.9, Identities: 24/26(92%), Gaps: 2/26(7%), Strand: Plus/Plus. |
Выдает находки с довольно хорошим показателем Ident и "лучшим из всех алгоритмов" E-value. |
| Discontiguous megablast | 1 | - | Не удалось обнаружить находку, которой бы не нашли другие алгоритмы |
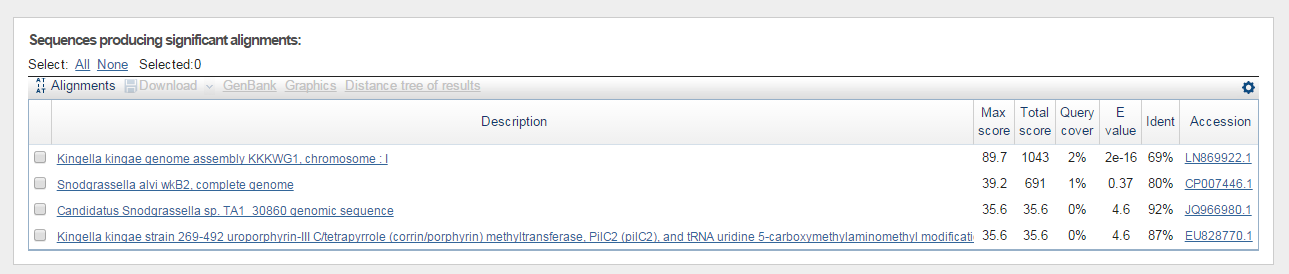
| Blastn | 4 | Candidatus Snodgrassella sp. TA1_30860 genomic sequence
Score: 35.6 bits(38), Expect: 4.6, Identities: 22/24(92%), Gaps: 0/24(0%), Strand: Plus/Plus. |
Выдает наибольшее число находок. Находки выдает со средними показателями по Ident, но не очень хорошим E-value. |
Рис. 3 - Результаты выдачи Megablast
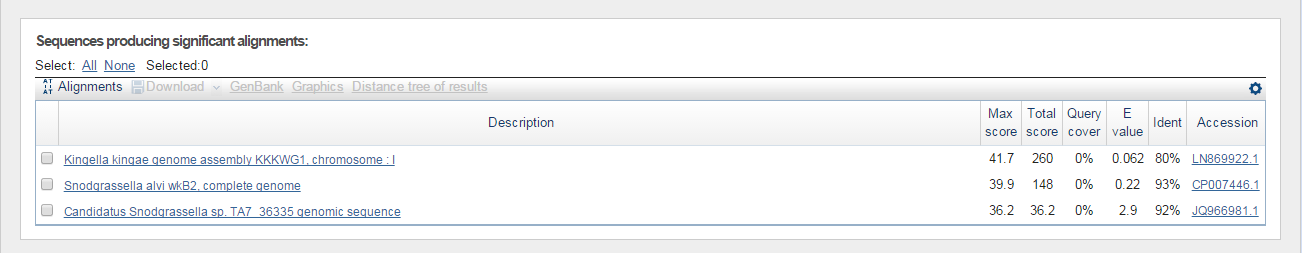 |
Рис. 4 - Результаты выдачи Discontiguous megablast
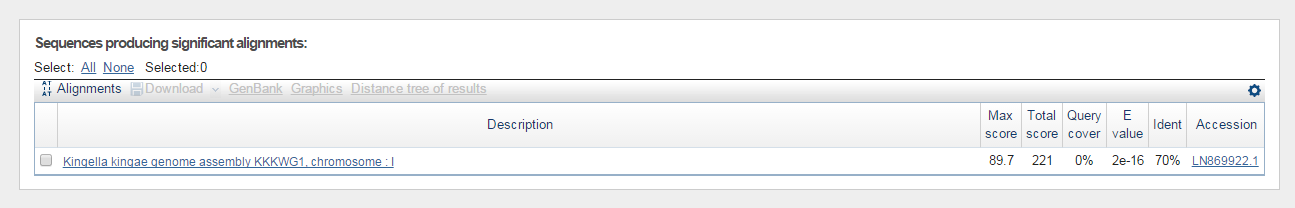 |
Рис. 5 - Результаты выдачи blastn
 |
| Вывод: Blastn выдает наибольшое число результатов, но у половины находок очень высокий E-value. Алгоритм Discontiguous megablast выдает самые лучшие данные по Max score из всех алгоритмов, его минус - наименьшее число находок. Megablast выдает хорошие результаты по Ident, средний Max score и относительно неплохой по сравнению с другими алгоритмами E-value. |
|
В этом задании необходимо было проверить наличие гомологов трех белков в геноме организма.
В качестве организма был взят Amoboaphelidium protococarum, сборка генома X5.
Были взяты следующие белки:
1) HSP7C_HUMAN - консервативный шаперон HSP70, белок теплового шока; 2) TERT_HUMAN - теломераза,восстанавливающая длину хромосомы при репликации; 3) PABP2_HUMAN - белок, cвязывающий поли(А) хвост матричной РНК. Сначала идет подготовка банка - команда (1). Для поиска был использован tblastn команда (2): |
|
(1) makeblastdb -in X5.fasta -dbtype nucl
(2) tblastn -query ---.fasta -db X5.fasta > ---.out (вместо --- ставится название белка). |
|---|
| Ссылки на файлы с последовательностями белков в формате fasta: HSP7C_HUMAN, PABP2_HUMAN, TERT_HUMAN. |
Таблица 3. Характеристика
| Белок | Число находок | Лучшая находка | Описание лучшей находки | Ответ (+,-) |
| HSP7C_HUMAN | 16 | scaffold-199 | Length = 1112851;
Score = 917 bits (2369), Expect = 0.0,
Method: Compositional matrix adjust. Identities = 474/607 (78%), Positives = 538/607 (89%), Gaps = 0/607 (0%), Frame = -2. |
"+": Таким образом, можно сделать вывод, что данный участок является гомологичным белку и выполняет сходные функции. |
| PABP2_HUMAN | 16 | E-value довольно неплох для некоторых находок, но Score не очень хороший. Поэтому в качестве лучшей была взята лучшая находка по Score-у: scaffold-100. | Length=762135, Score = 117 bits (292), Expect = 2e-28, Method: Compositional matrix adjust. Identities = 54/86 (63%), Positives = 64/86 (74%), Gaps = 0/86 (0%) Frame = +1. | "-": Скорее всего, не являются гомологами, так как для всех находок характерен невысокий Score, а для части из них еще и высокий E-value. |
| TERT_HUMAN | 3 | И E-value, и Score выдают не лучшие значения. Выбор "лучшего" производился по результатам Score: scaffold-17. | Length=2125590, Score = 105 bits (263), Expect = 8e-23, Method: Compositional matrix adjust. Identities = 151/568 (27%), Positives = 248/568 (44%), Gaps = 43/568 (8%) Frame = +1 | "-": Скорее всего, не являются гомологами, так как значения E-value и Score не являются удачными. |
|
В данном задании необходимо было найти один ген белка, закодированный в одном скэффолде "Amoeboaphelidium".
Необходио было выбрать один контиг длины десятков тысяч п.н.
Для выбора контига использовалась команда: infoseq X5.fasta -only -name -length.
Был выбран scaffold-59, длиной 17200.
Для получения последовательности была использована команда: seqret X5.fasta: scaffold-59 -out scaffold.fasta.
Ссылка на файл со скэффолдом Было проведено 2 поиска с ограничением по таксону Fungi: blastn и megablast. Поиск по megablast выдал всего 1 результат (рис. 1) с ident = 100%, e-value = 0.007, query cover = 0%. |
Рис. 6 - Megablast search
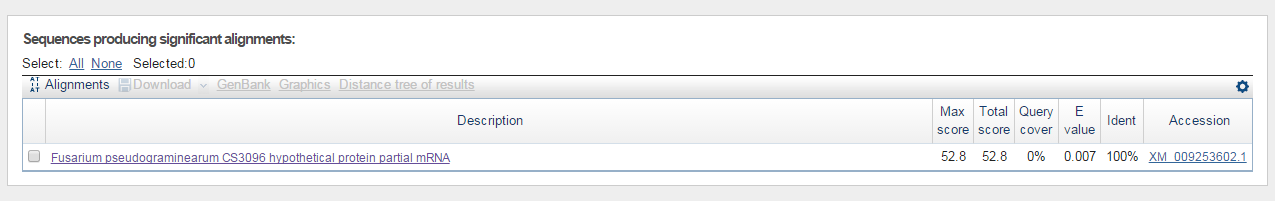 |
| На рис. 2 показана часть результатов по blastn поиску с ограничением по таксону Fungi. Как можно увидеть, довольно неплохой identity, query cover. На основании обоих поисков, можно сделать вывод, что закодирована в выбранном скэффолде гипотетическая частичная последовательность mRNA. |
Рис. 7 - Blastn search
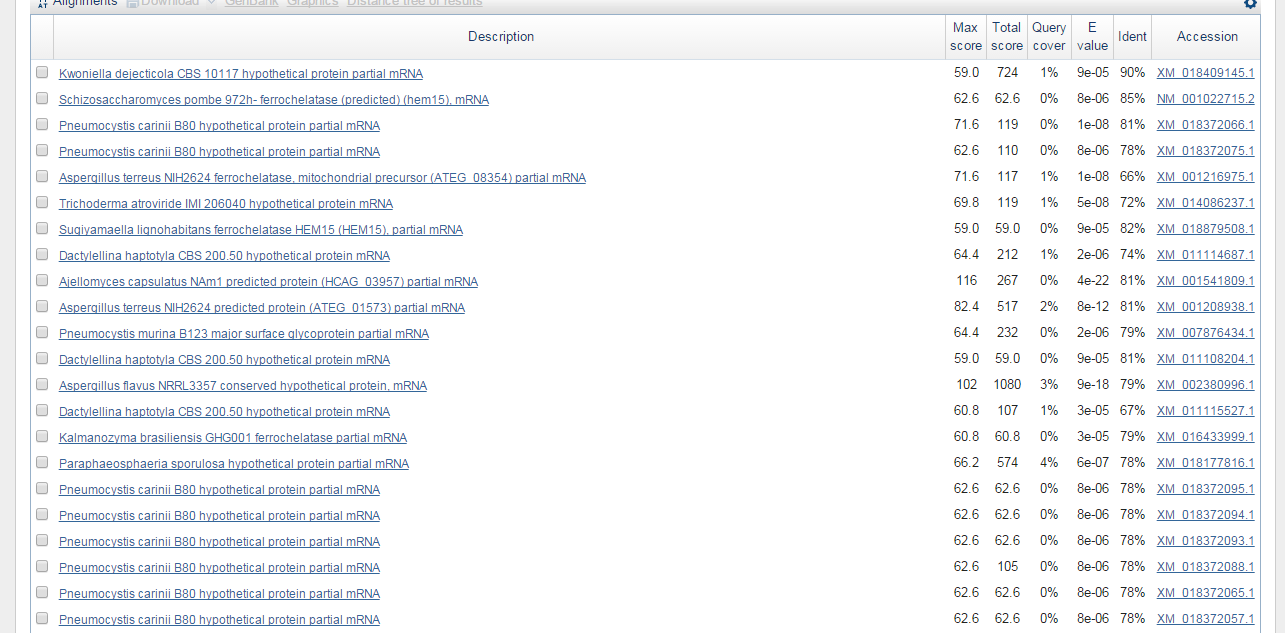 |
© Kalashnikova Anastasia, 2016