Реконструкция эволюции доменной архитектуры
Задание 1. Построить выравнивание представителей домена Pfam белков с разной доменной архитектурой
Выбор домена. В данном пункте необходимо было выбрать домен.
Я выбрала: KH domain (ID: KH_7; AC: PF17214).
Ссылка на страницу домена в Pfam.
Данный домен содержится в 204 последовательностях у 198 видов; для него известно 10 архитектур.
K гомологичный домен (KH domain) является эволюционно-консервативным примерно 70 кислотам,
данный домен широко представлен в самых разнообразных белках, связывающих нуклеиновые кислоты.
KH домен связывается с РНК или одноцепочечной ДНК и способен участвовать в узнавании РНК.
Также для данного домена известно 6 3D структур для 3-ех белков.
Ссылка на страницу Pfam с доменными архитертурами
JalView. Выравнивание было скачано из Pfam (Full):
в JalView: File > Fetch Sequences > Pfam (Full) > PF17214; и покрашено
ClustalX (By conservation: 30). Ссылки:
проект JalView;
выравнивание в fasta-формате.
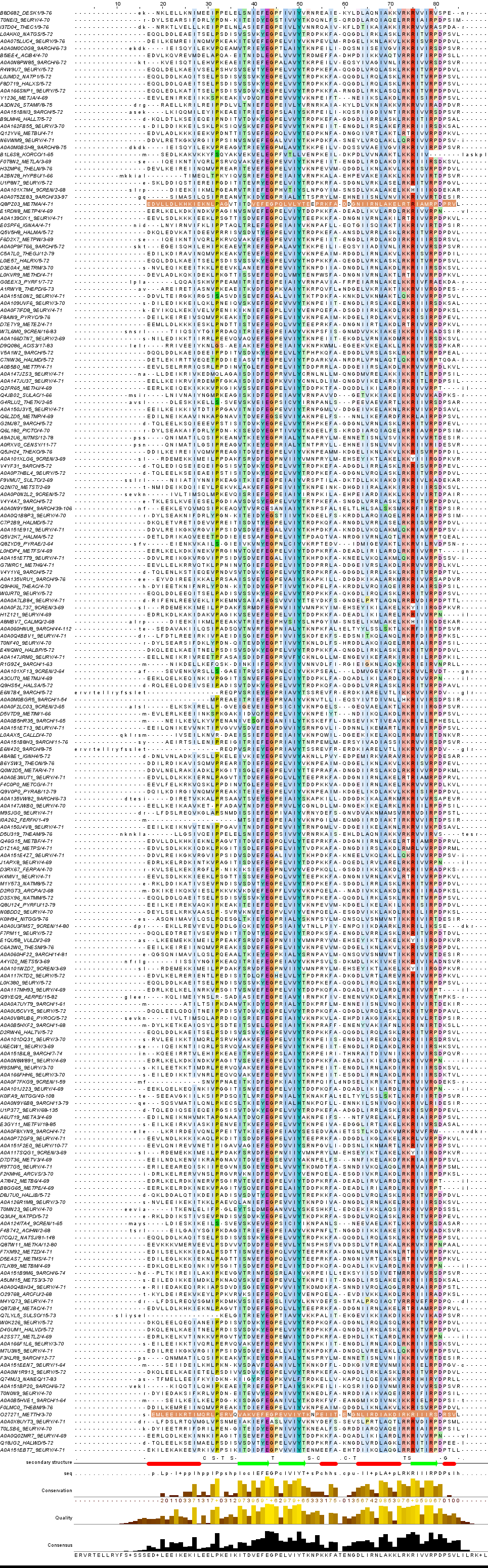
На рисунке 1 представлено изображение выравнивания.
Рис. 1 - Выравнивание последовательностей в раскраске Clustalx
 |
Выбор 2 архитектур, включающих мой домен. Мой домен включают в себя 10 архитектур, среди которых всего 4 архитектуры соответствуют требованию: существует не менее 20 последовательностей с данной архитектурой. Были выбраны две четырехдоменные архитектуры, информация по которым представлена в таблице 1.
Таблица 1. Выбор архитектур
| Номер архитектуры | Домены | Число представителей | Краткая характеристика других доменов |
| №1 | KH_7, Lactamase_B_6, Beta-Casp, RMMBL | 109 |
Beta-Casp: данный домен примыкает C-концом к beta-lactamase домену в эндонуклеазе,
процессирующей 3'-конец премРНК. Активный центр фермента располагается на стыке этих 2 доменов.
Lactamase_B и Lactamase_B_6: данные домены содержатся в основном в классе B бетта-лактамаз и и в некоторых других белках. Металло-бетта-лактамазы - важные фепменты, участвующие в процессе разрушения антибиотиков в антибиотик-резистентных бактериях. RMMMBL: Этот домен добавляет существенные структурные элементы в CASP-домен и является уникальным для РНК / ДНК-нуклеаз обработки, показывая, что они являются пре-мРНК-3'-процессирующими ээндонуклеазами. |
| №2 | KH_7, Lactamase_B, Beta-Casp, RMMBL | 27 |
Рис. 2 - Изображение архитектуры №1 - зеленым обозначен домен KH_7, желтым - RMMBL
 |
Рис. 3 - Изображение архитектуры №2 - зеленым обозначен домен KH_7, желтым - RMMBL
 |
Получение таблицы с информацией об архитектуре всех последовательностей. Файл swisspfam, содержащий эту информацию для всех последовательностей Uniprot, лежит в папке: /srv/databases/pfam/swisspfam.gz. Был использован скрипт swisspfam_to_xls.py, который отбирает последовательности с моим доменом и составляет Excel-таблицу - команда (1). Таким образом, был получен файл arc.xls. Далее необходимо было составить сводную таблицу: строки – AC последовательностей, столбцы – домены Pfam. В список последовательностей были добавлены колонки с информацией по таксономии: Uniprot > Retrieve > Cписок AC. Был получен файл list.txt, который был подан на вход питону: скрипт uniprot_to_taxonomy.py; команда (2). Полученная таксономия была перенесена в основную таблицу. Также был добавлен лист с длинами выбранных доменов из каждой последовательности.
|
(1) python swisspfam-to-xls.py -z -i swisspfam.gz -p PF17214 -o arc.xls
(2) python uniprot-to-taxonomy.py -i list.txt -o taxonomy.xls |
|---|
Выбор таксона и подтаксона. В качестве таксона: Archaea. Euryarchaeota не делится на подтаксоны, состоящие из > 20 последовательностей, поэтому было принято решение за один подтаксон принять Euryarchaeota (E), а за другой (условно) - совокупность представителей Crenarchaeota и Thaumarchaeota (CT). Далее необходимо было выбрать последовательности так, чтобы в каждом подтаксоне от каждой архитектуры было по 20 последовательностей. С первой архитектурой не возникло проблем - она довольно распространена. А вот найти 20 последовательностей с 2-ой архитектурой оказалось невозможным: нашлось 7 последовательностей для E, 6 - для CT. Недостаток компенсировался последовательностями 1 архитектуры. Ссылка на итоговую таблицу.
Таблица 2. Поиск подтаксонов
| Название подтаксона | Число представителей |
| Euryarchaeota | 76 |
| Crenarchaeota | 18 |
| Thaumarchaeota | 6 |
| Candidatus Bathyarchaeota/environmental samples/not stated | 25 |
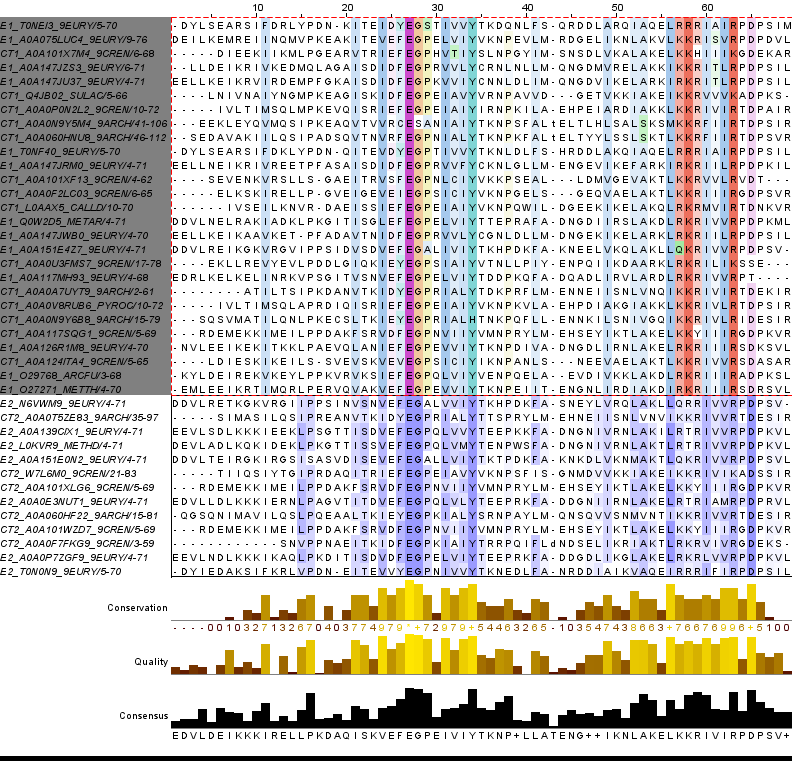
Последовательности выбранных представителей примерно одинаковы по длине доменов, поэтому особых проблем возникнуть не должно. В выравнивании были оставлены только выбранные последовательности. Для этого: был получен файл с id выбранных последовательностей (ссылка). Далее с помощью команды (1) было получено выравнивание моих последовательностей, к id которых я добавила подтаксон и архитектуру (CT[1,2];E[1,2]). Полученное выравнивание в fasta-формате. Выравнивание было открыто в JalView: были убраны пустые колонки (Edit > Remove Empty Columns), были созданы группы (по архитектурам: Selection > Create Group > Edit name and description), также я подчистила N,C-участки и отсортировала по группе (Calculate > Sort). 1-ая группа была покрашена ClustalX (conservation: 30), 2-ая - BLOSUM62 (conservation: 30) (рис. 4). Особо выбивающихся последовательностей я не нашла, поэтому ничего удалять не стала. Ссылка на проект JalView.
В выравнивании группы 1 можно четко выделить 2 блока консервативных/полуконсервативных колонок. Не может не радовать тот факт, что эти блоки можно относительно соотнести с блоками выравнивания группы 2. Присутствует несколько консервативных колонок, которые были бы абсолютно консервативны, если бы не пара последовательностей; абсолютно консервативные колонки. На мой взгляд, выравнивание приемлимое, поэтому строить дерево можно.
Рис. 4 - Выравнивание последовательностей
 |
| (1) python filter-alignment.py -i Domain_1.fasta -m id.txt -o pr11.fasta -a "_" |
|---|
Задание 2. Построение филогенетического дерева домена
Было построено дерево по полученному в предыдущем задании выравниванию.
Обозначения: подтаксоны: Euryarchaeota (E), Crenarchaeota и Thaumarchaeota (CT);
архитектуры: 1, 2 (в соответствии с таблицей 1).
Таким образом, обозначение каждой последовательности: подтаксон(архитектура)_ID.
Для построения дерева были использованы: программа MEGA; метод NJ; бутстреп (100 реплик).
Скобочная структура дерева представлена здесь.
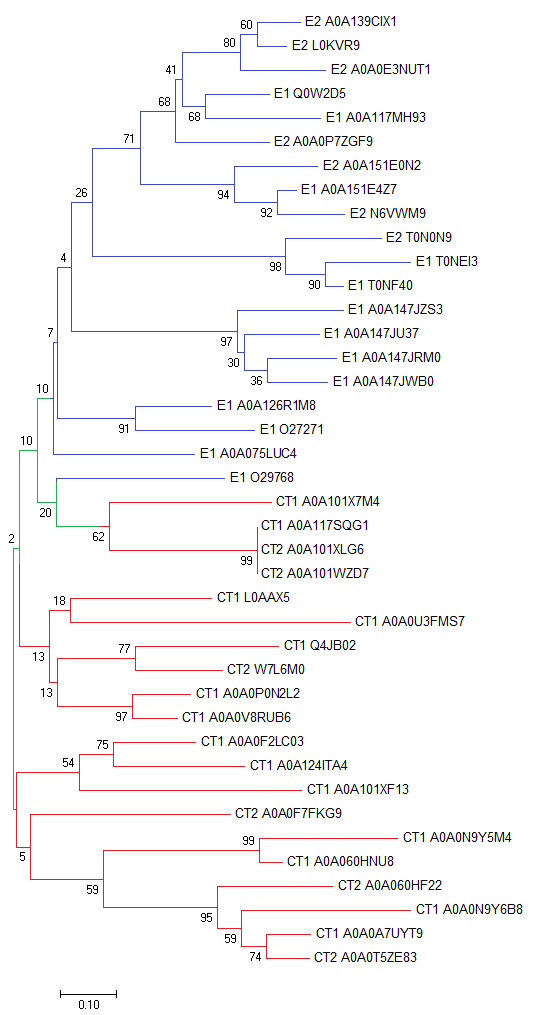
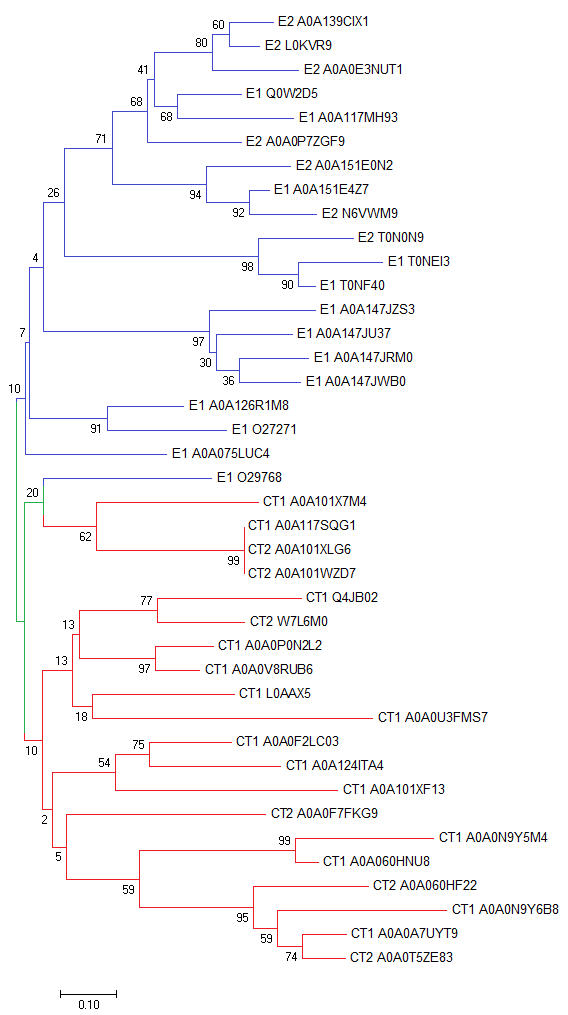
MEGA построила дерево представленное на рисунке 5.1.
Однако мне не понравилось выбранное укоренение, и я решила переукоренить сама.
В переукорененном мной дереве есть более четкое деление на таксоны
(из него выделяется всего 1 последовательность подтаксона E) - рисунок 5.2.
Какого-то четкого разделения на архитектуры в подтаксонах не наблюдается,
откуда можно сделать вывод, что нельзя точно предсказать какие именно эволюционные события происходили.
Рис. 5 - Дерево до и после переукоренения - увеличение при нажатии.
Синий цвет - ветви, все листья которых принадлежат подтаксону E; красный цвет - подтаксону CT.
 |
 |