Определение сайтов связывания данного транскрипционного фактора в данном участке хромосомы человека
Подготовка чтений
Файлы .fastq с ридами Illumina, полученные в результате ChIP-seq эксперимента,
разделены на отдельные файлы, соответствующие участкам хромосом.
Мне достался файл chipseq_chunk49.fastq, который лежит в папке
/P/srv/databases/ngs/chipseq_y14.
Файл был перенесен в /nfs/srv/databases/ngs/asya.kalashnikova/49.fastq.
Далее, как и в прошлом семестре, был осуществлен контроль качества прочтений.
Была использована команда (1) при работе с программой FastQC.
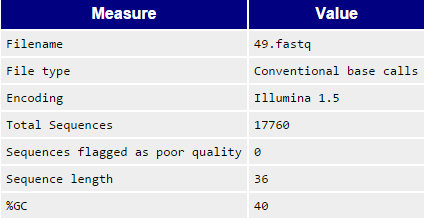
В итоге были получены 2 файла:
49_fastqc.html и 49_fastqc.zip.
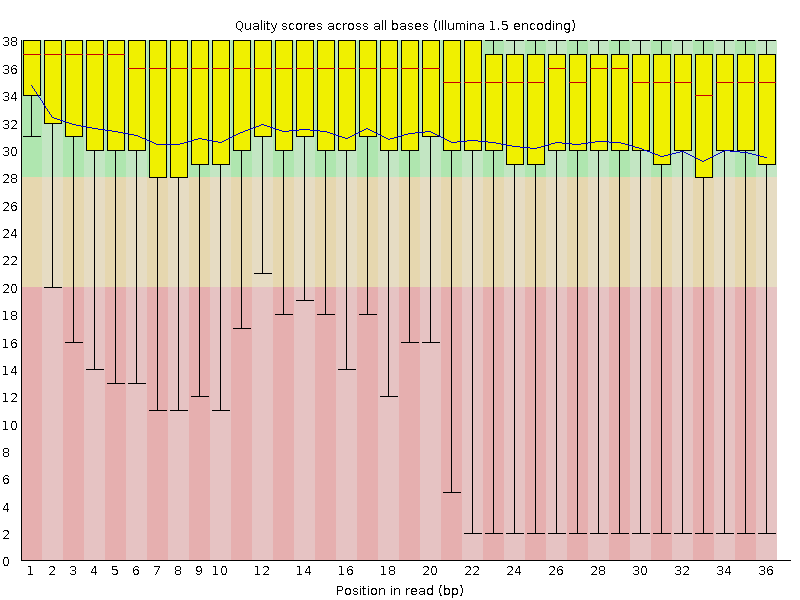
Хоть FastQC и убеждает, что чтений с плохим качеством не обнаружилось,
линии на "Per base sequence quality" оказались слишком низкими,
поэтому я решила почистить чтения программой Trimmomatic с помощью команды (2).
Остались чтения длиной больше 36, причем с конца каждого чтения были отрезаны нуклеотиды с качеством ниже 40.
Далее был проведен анализ очищенных чтений, используя команду (3).
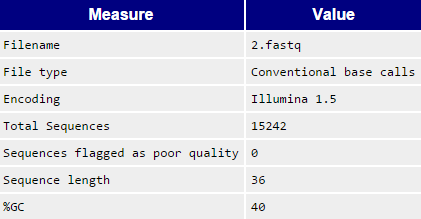
Получены 2 файла:
2_fastqc.html и 2_fastqc.zip.
Как можно увидеть на рис.1 очистка существенно улучшила показатель качества ридов.
|
(1) fastqc 49.fastq
(2) java -jar /usr/share/java/trimmomatic.jar SE -phred33 49.fastq 2.fastq TRAILING:40 MINLEN:36 (3) fastqc 2.fastq |
|---|
Рис. 1 - Per base sequence quality (до и после чистки) - увеличение при нажатии
 |
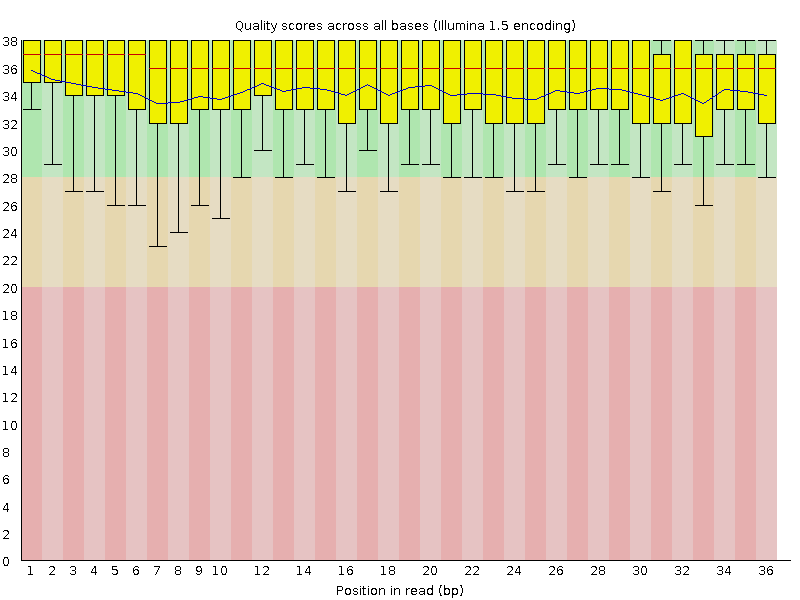 |
Рис. 2 - Basic Statistics (до и после чистки) - увеличение при нажатии
 |
 |
Картирование чтений
Необходимо было откартировать чтения с помощью программы BWA.
Я воспользовалась проиндексированным геномом человека, который лежал в папке /srv/databases/ngs/hg19.
Картирование осуществлялось с помощью команды (1).
Далее происходил анализ выравнивания:
сначала выравнивание было переведено в бинарный формат bam с помощью пакета
samtools, команды (2). Опция -o была использована для указания выходного файла,
-b - для смены формата с sam на bam.
Получившийся файл был отсортирован
по координате в референсе начала чтения - команда (3),
опция -T была использована для записи временного файла chip, в который выводится stdout).
Далее файл sort.bam был проиндексирован командой (4) и исследован
на предмет числа откартированных чтений на геном с помощью команды (5).
Был получен файл kart.txt (ссылка).
Число не картированных/картированных чтений на хромосому: 36/15206 из 15242.
Из 15206 картированных чтений 14164 приходится на 10-ую хромосому,
поэтому именно прочтения с 10-й хромосомы я и буду использовать для анализа.
|
(1) bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa
2.fastq > kart.sam
(2) samtools view kart.sam -b -o kart.bam (3) samtools sort kart.bam -T chip -o kart.sorted.bam (4) samtools index kart.sorted.bam (5) samtools idxstats kart.sorted.bam > kart.txt |
|---|
Поиск пиков
Для поиска пиков (peak calling) была использована программа MACS и команда (1), так как при попытке использования более простой команды (без nomodel), программа выдает, что пиков слишком мало. Опция -n служит для изменения названия эксперимента. Получены файлы, содержащие информацию о найденных пиках: chip10_peaks.narrowPeak, chip10_peaks.xls и chip10_summits.bed. На моей хромосоме программа нашла 9 пиков. В таблице 1 и на рисунке 3 представлена информация о найденных пиках.
| (1) macs2 callpeak -n chip10 -t chipseq_chunkX.sorted.bam --nomodel |
|---|
Таблица 1. Описание найденный пиков
| Номер пика | Координата начала | Координата конца | Длина | Расстояние вершины от старта пика | -Log10 (pvalue) | -Log10 (qvalue) |
| №1 | 34386711 | 34386980 | 270 | 113 | 29.39742 | 22.16956 |
| №2 | 34410299 | 34410627 | 329 | 164 | 23.88743 | 16.88431 |
| №3 | 34505429 | 34505666 | 238 | 78 | 12.99871 | 6.54739 |
| №4 | 34543636 | 34543835 | 200 | 108 | 16.89913 | 10.23025 |
| №5 | 34608523 | 34608786 | 264 | 112 | 13.44860 | 6.97747 |
| №6 | 34634307 | 34634671 | 365 | 153 | 22.70822 | 15.76915 |
| №7 | 35025949 | 35026271 | 323 | 149 | 43.55650 | 34.07337 |
| №8 | 35623334 | 35623609 | 276 | 114 | 13.54712 | 7.07343 |
| №9 | 35974340 | 35974572 | 233 | 136 | 12.77258 | 6.34060 |
С помощью UCSC Genome Browser (My Data > Custom Tracks) была произведена визуализация пиков (рис. 3). На вход подавалось: "track type=narrowPeak visibility=3 db=hg19 name="Peaks" description="NarrowPeak chr10" browser position chr10:34386711-35974572" в начале файла chip10_peaks.narrowPeak.
Рис. 3 - Визуализация пиков - увеличение при нажатии
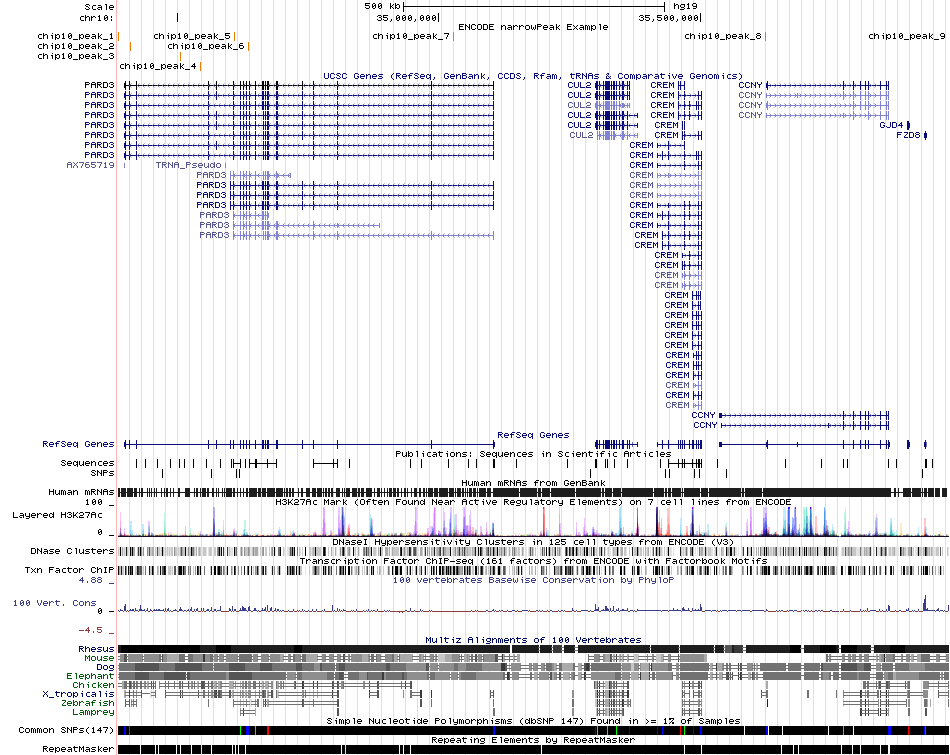 |
Далее необходимо было изучить несколько пиков в более крупном масштабе. Достоверность пиков можно оценить как по q-value, так и по p-value. В выданном файле указан -log10(p-value). Чем больше это значение, тем меньше десятичный логарифм (следовательно, p-value). Я бы сказала, p-value у всех моих пиков достаточно низкий, что позволяет говорить об их достоверности. Для более подробного изучения были выбраны пики 1 и 7, так как у них наилучший p-value.
Рис. 4 - Визуализация пика 1 - увеличение при нажатии
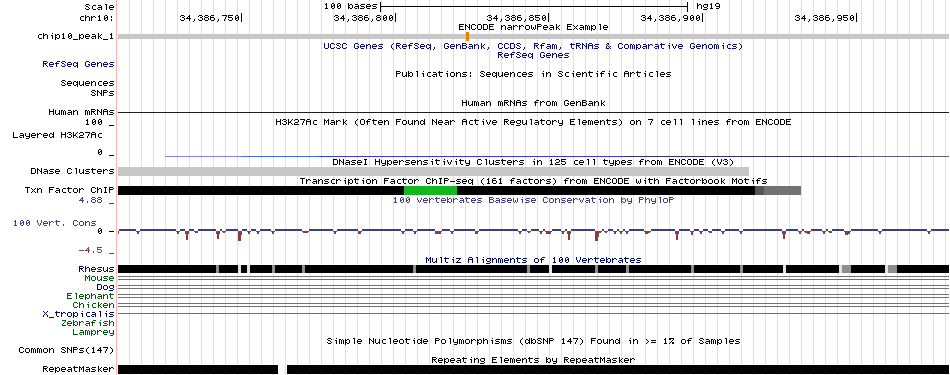 |
Рис. 5 - Визуализация пика 7 - увеличение при нажатии
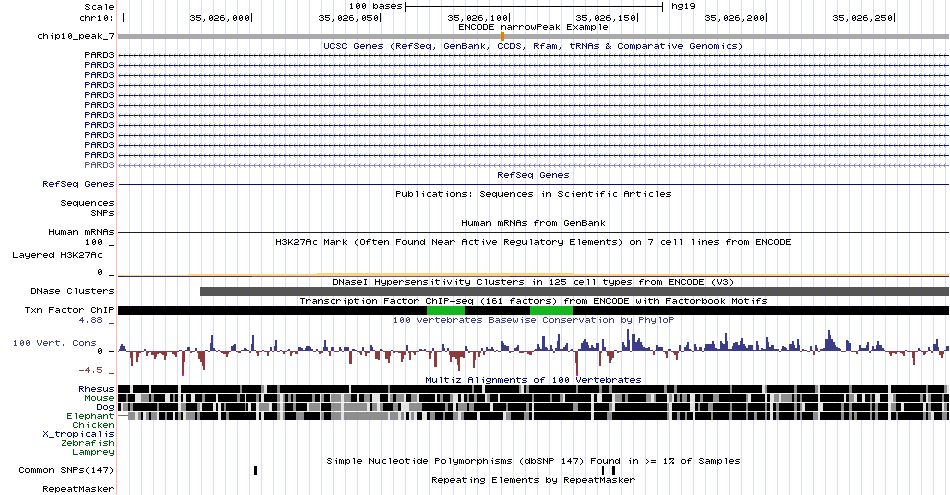 |
Как можно заметить, пик №1 не пересекается ни с какими функциональными элементами генома, в то время как пик №7 картируется на ген PARD3. Данный ген кодирует представителя PARD семейства белков. Белки данного семейства способны взаимодействовать друг с другом и с другими белками. Они влияют на асcиметричное деление клетки.