Подготовка чтений
Перед началом работы была создана директория /nfs/srv/databases/ngs/atitova, где затем и велась дальнейшая работа.
Анализ качества чтений
Команда
[1]:
fastqc chr17.fastqc
Результат:
chr17_fastqc.html
Очистка чтений
Команда
[2]:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr17.fastq chr17_trimmomatic.fastq TRAILING:20 MINLEN:50
Результат:
chr17_trimmomatic_fastqc.html
На рисунках 1 и 2, расположенных ниже, вы можете видеть графики качества чтения, полученные с помощью программы fastqc. Синяя линия соответсвует среднему качеству чтения,
красная линия - медиане, желтые столбики - разнице между верхним и нижним квартилями.
Поле графика поделено на три области - зеленую, желтую и красную, попадание в которые разных элементов графика позволяет судить о качестве их
прочтения.
По графикам видно, что качеcтво прочтения улучшилось после работы программы trimmomatic - практически все элементы содержатся в зеленой области.
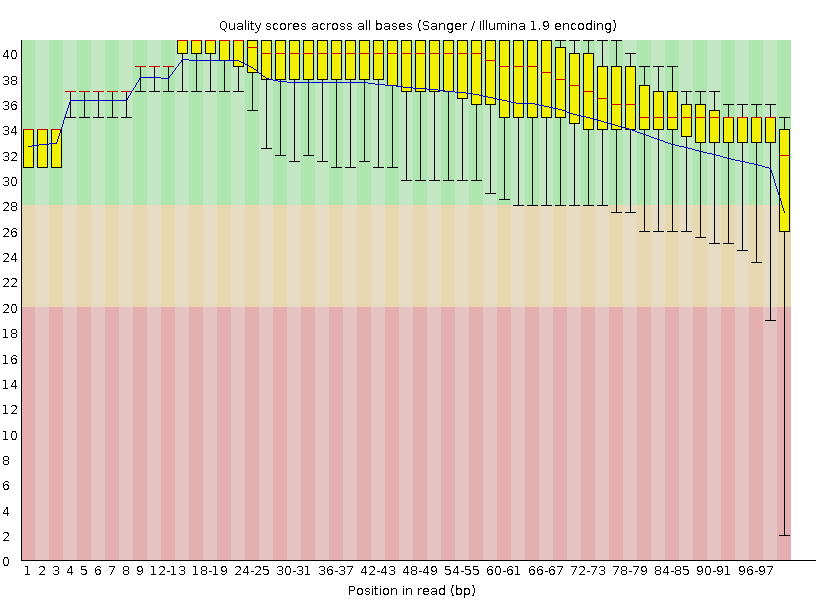 |
| Рис. 1. Per base quality до чистки |
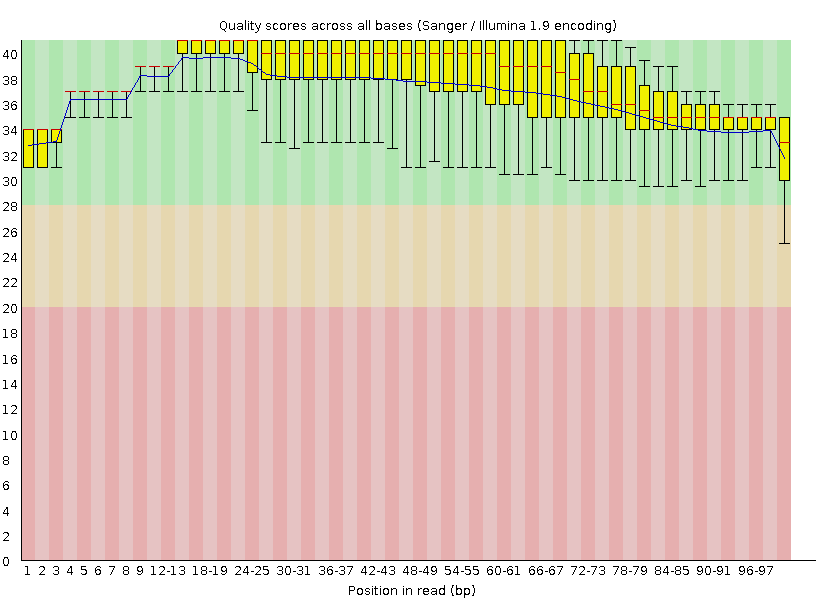 |
| Рис. 2. Per base quality после чистки |
 |
| Рис. 3. Ящик с усами / диаграмма размахов / boxplot. |
Картирование чтений
Картирование чтений
Команда, вызывающая программу:
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
Команда, индексирующая референсную последовательность и выдающая множество файлов с расширением .ht2
[3]:
hisat2-build chr17.fasta chr17
Команда, которая строит выранивание прочтения и референса
[3]:
hisat2 -x chr17 -U chr17_trimmomatic.fastq --no-spliced-alignment --no-softclip > align.sam
Для получения файлов без разрывов и подрезания прочтений были использованы:
"--no-softclip", соответственно.
Результат:
align.sam
В результате работы hisat2:
- не были выравнены: 34 прочтения;
- были выравнены один раз: 7109 прочтений;
- были выравнены больше одного раза: 3725 прочтений.
Анализ выравнивания
Команда, переводящая выравние с референсом в бинарный формат
[4]:
samtools view align.sam -bo align.bam
Команда, сортирующая выравнивание чтений с референсом по координате в референсе начала чтения.:
samtools sort align.bam -T myfile.txt -o sort_align.bam:
Команда, индексирующая отсортированный файл .bam:
samtools index sort_align.bam
И далее:
samtools idxstats sort_align.bam.bai > idxstats.txt.
Результат:
idxstats.txt
Анализ SNP
Поиск SNP и инделей
Команда, создающая файл с полиморфизмами на основе референсной последовательности и файла с выравниванием прочтений:
samtools mpileup -uf chr17.fasta sort_align.bam > snp.bcf
Команда, создающая список отличий между референсом и чтениями:
bcftools call -cv snp.bcf > snp.vcf
Результат:
snp.vcf
В результе работы программы bcftools было найдено 58 полиморфизмов, из которых 4 инделя и 54 замены.
| Таблица 1. Описание трех полиморфизмов из файла .vcf |
| Хромосома |
Координата |
Тип полиморфизма |
Референс |
Прочтение |
Глубина |
Качество |
| chr17 | 44753827 | Замена | G | A | 1 | 11.3429 |
| chr17 | 79534241 | Делеция | ATCTTTCT | ATCT | | 13.657 |
| chr17 | 79596811 | Замена | C | T | 48 | 221.999 |
Аннотация SNP
Команда, создающая список отличий между референсом и чтениями:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/atitova/snp.vcf >
/nfs/srv/databases/ngs/atitova/snp.avinput
Результат:
snp.avinput.
Далее этот файл использовался при поиске
Аннотация по dbsnp
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/atitova/rs.snp -build hg19 -
dbtype snp138 /nfs/srv/databases/ngs/atitova/snp.avinput /nfs/srv/databases/annovar/humandb/
В результате из 58 полиморфизмов:
Аннотация по refgene
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -out /nfs/srv/databases/ngs/atitova/rs.refgene -build hg19
/nfs/srv/databases/ngs/atitova/snp.avinput /nfs/srv/databases/annovar/humandb/
В результате было получено два файла, содержащие информацию о полиморфизмах:
В первом содержится информация о полиморфизмах, сортированных в соответствии с их положением в геноме. Ниже вы можете видеть таблицу, которая это
описывает.
| Таблица 2. |
| intronic |
exonic |
intergenic |
UTR3 |
| 49 |
3 |
1 |
5 |
На основе данных из таблицы можно заключить, что наибольшее количество замен приходится на интроны. Возможно, это объясняется тем, что эти замены
не влияют на продукты геном и, как следствие, не подвержены отбору.
Во втором содержится информация о заменах в экзонах. Ниже вы можете видеть таблицу, содержащую информацию о изменениях в экзонах гена
[5], кодирующего
фактор распознавания убиквитина. ( Убиквитин - небольшой консервативный белок эукариот, участвующий в регуляции процессов внутриклеточной деградации других белков,
а также их функций.[6] )
| Таблица 3. |
| Координаты |
Ген |
Экзон |
Тип замены |
Качество прочтения |
Глубина прочтения |
Замена |
| 79589242 - 79589242 |
NPLOC4 |
exon3 |
Синонимичная |
221.999 |
103 |
G -> A |
| 79596811 - 79596811 |
NPLOC4 |
exon2 |
Синонимичная |
221.999 |
48 |
C -> T |
Аннотация по Clinvar
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/atitova/rs.clinvar -dbtype
clinvar_20150629 -buildver hg19 /nfs/srv/databases/ngs/atitova/snp.avinput /nfs/srv/databases/annovar/humandb/
Результатом являются два файла:
rs.clinvar.hg19_clinvar_20150629_dropped
,
rs.clinvar.hg19_clinvar_20150629_filtered
Первый файл (rs.clinvar*dropped) оказалася пустым. Это означает, что из всех snp не нашлось ни одного, имеющего клиническое значение. Второй файл содержит
информацию о неаннотированных snp.
Аннотация по 1000Genomes
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/atitova/rs.1000g -buildver hg19
-dbtype 1000g2014oct_all /nfs/srv/databases/ngs/atitova/snp.avinput /nfs/srv/databases/annovar/humandb/
Поиск по 1000Genomes дает информацио о частоте встречаемости анализируемых полиморфизмов:
- самая высокая - 0.970847;
- самая низкая - 0.000199681.
Аннотация по GWAS
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/atitova/rs.gwas -build hg19
-dbtype gwasCatalog /nfs/srv/databases/ngs/atitova/snp.avinput /nfs/srv/databases/annovar/humandb/
Поиск по этой GWAS дает информацию о связи полиморфизмов с фенотипическими проявлениями. В результате работы этой команды был получен пустой файл,
а это значит, что не нашлось ни одного фенотипического проявления.
Сводная таблица по базам данных
По это ссылке доступна сводная таблица по базам данных.
Источники:
[1]: FastQC
[2]: Trimmomatic
[3]: Hisat2
[4]: Annovar
[5]: NCBI / GENE: NPLOC4 NPL4 homolog, ubiquitin recognition factor [ Homo sapiens (human) ]
[6]: Википедия. Убиквитин
