Перед началом работы были скачаны файлы с одноконцевыми чтениями chr17.1.fastqc и chr17.2.fastqc из /P/y14/term3/block4/SNP/rnaseq_reads в директорию
из предыдущего практикума (/nfs/srv/databases/ngs/atitova). В этой же директории и велась вся дальнейшая работа с данными файлами. Работа велась с
обеими биологическими репликами.
Подготовка чтений
Анализ качества чтений
Команда:
fastqc chr17.1.fastqc
fastqc chr17.2.fastqc
Результат:
Ниже вы можете видеть изображения Per base quality для chr17.1 и chr17.2.
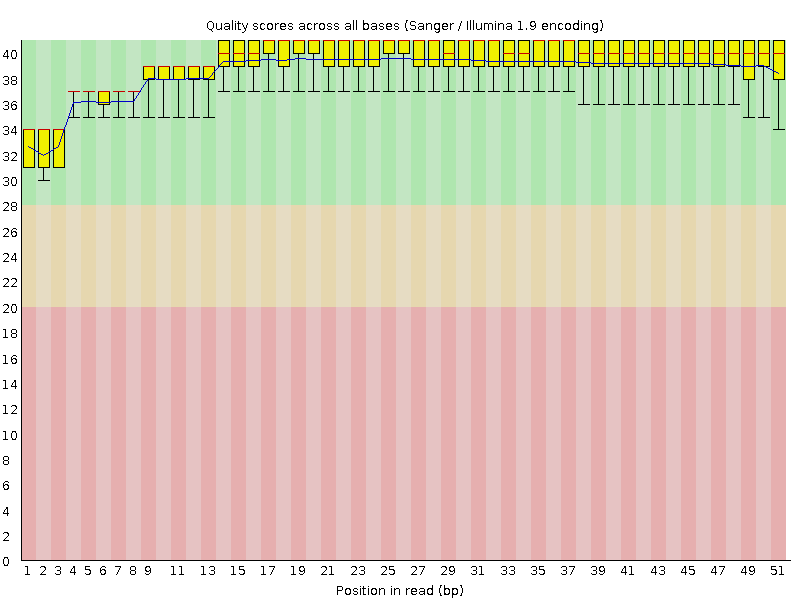 |
| Рис. 1. Per base quality chr17.1 |
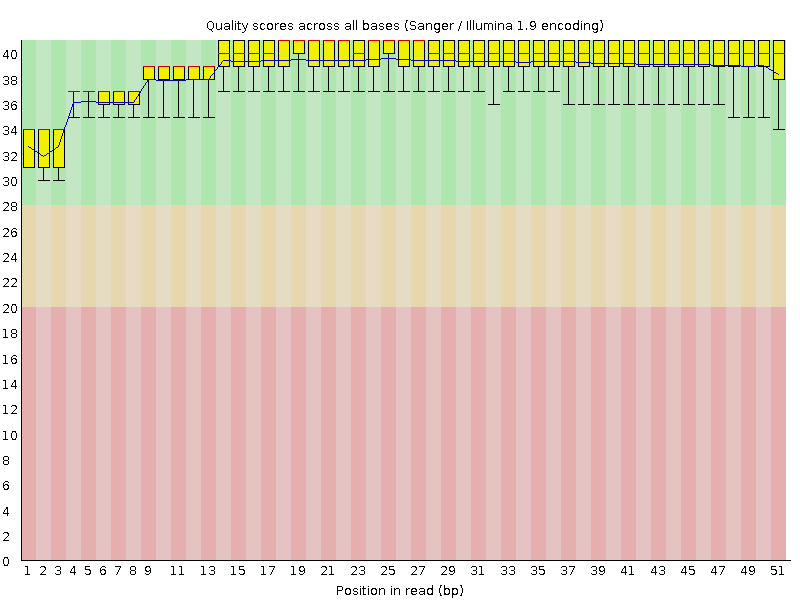 |
| Рис. 2. Per base quality chr17.2 |
На основе полученных отчетов можно заключить, что обе реплики не нуждаются в последующей чистке: все прочтения лежат в зеленой зоне,
длина прочтений chr17.1 составляет 25-51, chr17.2 - 32-51.
Картирование чтений
Картирование чтений
В результате выполнения практикума 11 уже имеется проиндексированная референсная последовательность.
Команда, вызывающая программу:
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
Команда, индексирующая референсную последовательность и выдающая множество файлов с расширением .ht2 (взят результат выполнения этой команды из 11
практикума):
hisat2-build chr17.fasta chr17
Команда, которая строит выранивание прочтения и референса:
hisat2 -x chr17 -U chr17.1.fastq --no-softclip > align1.sam
hisat2 -x chr17 -U chr17.1.fastq --no-softclip > align1.sam
В отличии от предыдущего практикума при выполнении этого задания был убран параметр "--no-spliced-alignment". Это было сделано из-за того, что работа
велась с последовательностями РНК, прошедшими вырезание интронов, и поэтому в них могут быть разрывы (относительно последовательнсти генома).
Результаты:
В результате:
Для первой реплики: откартировано 10407 прочтений, из которых 329 не были выравнены,
10001 были выравнены один раз, 77 были выравнены больше одного раза;
Для второй реплики: откартировано 8195 прочтений, из них 292 не были выравнены, 7847 были выравнены
один раз, 56 были выравнены больше одного раза.
Анализ выравнивания
Команда, переводящая выравние с референсом в бинарный формат:
samtools view align1.sam -bo align1.bam
samtools view align2.sam -bo align2.bam
Команда, сортирующая выравнивание чтений с референсом по координате в референсе начала чтения:
samtools sort align1.bam -T myfile.txt -o sort_align1.bam
samtools sort align2.bam -T myfile.txt -o sort_align2.bam
Команда, индексирующая отсортированный файл .bam:
samtools index sort_align1.bam
samtools index sort_align2.bam
Подсчет прочтений
Эта часть практикума выполнялась с помощью программы bedtools[1].
Команда, вызывающая bedtools:
export PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin
Команда, переводящая файл формата .bam в формат .bed и записывающая в файл 1.bed:
bedtools bamtobed -i sort_align1.bam > 1.bed
Команда, пересекающая общий файл с разметкой генов с чтениями. (Параметр -u оставляет только те чтения, которые хотя бы раз пересеклись с генами из
общего файла.)
bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b 1.bed -u > 2.bed
Результат:
2.bed
| Таблица 1. Итоги работы программы bedtools |
| Хромосома |
Ген |
Тип гена |
Количество прочтений |
| chr17 |
RP11-580I16.2 |
antisense |
6 |
| chr17 |
KPNB1[2] |
protein_coding |
306 |
| chr17 |
RP11-138C9.1 |
antisense |
3 |
bedtools
1. Получите из файла в выравниванием файл с чтениями в формате fastq
Команда, переводящая файл с выравниванием формата .bam в файл формата .fastq:
bedtools bamtofastq -i sort_align1.bam -fq sort_align1.fq
Результат:
sort_align1.fq
2.Получите файл с нуклеотидной последовательностью (.fasta) для одного из покрытых Вашими чтениями генов.
Команда:
bedtools getfasta -fi chr17.fasta -bed gene_1.bed -fo gene_1.fasta -name
Результат:
gene_1.fasta, файл
gene_1.bed
4. Объедините Ваши чтения в кластеры (используйте bed файл с выровненными чтениями из Обязательной части задания).
Команда, объединяющая прочтения в кластеры:
bedtools cluster -i 2.bed -s > cluster.bed
Результат:
cluster.bed (в этом файле список прочтений с указанием к какому
кластеру какое прочтение относится. Всего получилось 20 кластеров.)
