Задание 1. Предсказание вторичной структуры заданной тРНК
Упражнение 1. Предсказание вторичной структуры тРНК путем поиска инвертированных повторов
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях.
С ее помощью были найдены возможные комплементарные участки в последовательности исследуемой тРНК.
Результат работы этой программы вы можете видеть ниже. Красным цветом выделены те нуклеотиды, которые совпали с результатами, полученными ранее с
помощью find_pair.
SEQUENCE: Score 71: 22/31 ( 70%) matches, 6 gaps
1 ggga-t-t-tagctcagttggga-gagcgccaga-ct 32
|||| | | | | || | | || | ||||| ||
70 ccctaagacacctag-cttgtgtcctggaggtctaga 35
Упражнение 2. Предсказание вторичной структуры тРНК по алгоритму Зукера.
Программа RNAfold из пакета Viena Rna Package реализует алгоритм Зукера. На вход программа получает последовательность тРНК в FASTA - формате, а на выход
выдает последовательность, в которой "(" и ")" соответствуют 5'- и 3'-основаниям соответственно, а "." - неспаренное основание. Ниже вы можете видеть
результ работы этой программы.
>5AXM:P|PDBID|CHAIN|SEQUENCE
GGGAUUUAGCUCAGUUGGGAGAGCGCCAGACUGAAGAUCUGGAGGUCCUGUGUUCGAUCCACAGAAUCCCCACCA
((((((..((((........)))).(((((.......))))).....(((((.......)))))))))))..... (-21.90)
В моем случае программа с первого раза довольно точно определила 3 стебля и почти полностью 4-й. А ниже вы можете видеть изображение, полученное
с помощью сервиса RNAfold.
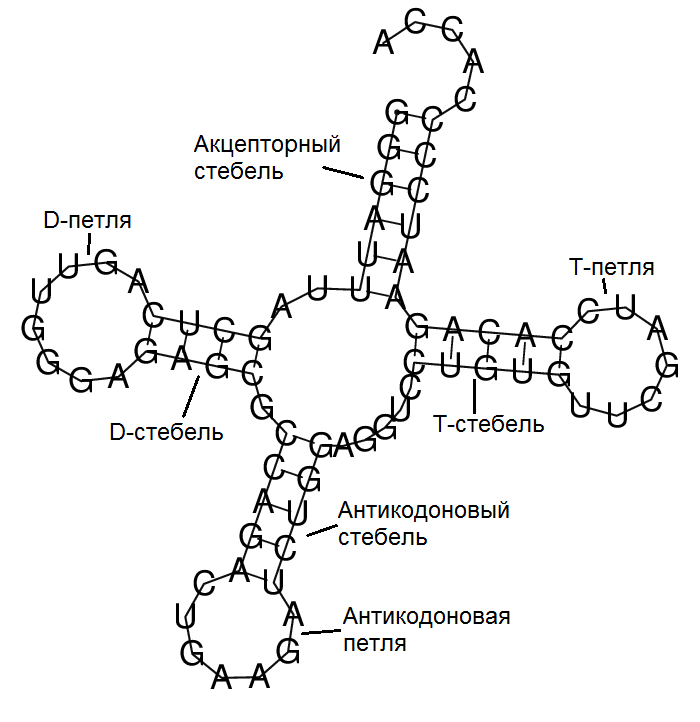 |
| Рис. 1. Предсказанная с помощью RNAfold структура РНК |
Сравнение трех полученных результатов вы можете видеть в таблице, приведенной ниже.
| Таблица 1. Реальная и предсказанная вторичная структура тРНК из файла 5axm.pdb |
| Участок структуры |
Позиции в структуре (по результатам find_pair) |
Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель |
5'-2-71-3'
5'-7-66-3'
Всего 6 пар
|
4/6 |
6/6 |
| D-стебель |
5'-10-25-3'
5'-13-22-3'
Всего 4 пары
|
0/4 |
4/4 |
| T-стебель |
5'-49-65-3'
5'-53-61-3'
Всего 5 пар
|
0/5 |
5/5 |
| Антикодоновый стебель |
5'-38-32-3'
5'-44-26-3'
Всего 7 пар
|
5/7 со сдвигом на один |
5/7
Такой результат верояно получен из-за неканонического взаимодействия пар G-A и A-C.
|
| Общее число канонических пар нуклеотидов |
22 |
10 |
20 |
Очевидно, с программой RNAfold были получены более достоверные результаты (20 правильно предсказанных пар, против 10 у find_pair).
Задание 2. Поиск ДНК-белковых контактов в заданной структуре
Упражнение 1
Для структуры с PDB_ID 4Z1X в JMol были заданы следующие множества:
- Множество атомов кислорода 2'-дезоксирибозы (set1).
- Множество атомов кислорода в остатке фосфорной кислоты (set2).
- Множество атомов азота в азотистых основаниях (set3).
Воспользовавшись кнопками, расположенными под апплетом, вы можете управлять скриптом ("Start script" для запуска, "Resume" для продолжения
выполнения скрипта, "Reset" - вернуться к исходному изображению),
последовательно отображающим данные множества атомов в проволочной модели ДНК.
Ссылка на скрипт
Упражнение 2
В данном упражнении необходимо было описать ДНК-белковые контакты в заданной структуре, а также сравнить количество контактов разной природы.
При этом мы считаем полярными атомы кислорода и азота, а неполярными атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние
между полярным атомом белка и полярным атомом ДНК меньше 3.5. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии
меньше 4.5.
Было определено число контактов и заполнена таблица, которую вы можете видеть ниже.
| Таблица 2. Контакты разного типа в комплексе 4z1x.pdb |
| Контакты атомов белка с |
Полярные |
Неполярные |
Всего |
| остатками 2'-дезоксирибозы |
3 |
53 |
56 |
| остатками фосфорной кислоты |
29 |
19 |
48 |
| остатками азотистых оснований со стороны большой бороздки |
15 |
66 |
81 |
| остатками азотистых оснований со стороны малой бороздки |
0 |
10 |
10 |
На основе данных второй таблицы можно сделать предоложение о том, что белок в основном взаимодействует с сахаро-фосфатным остовом ДНК (фосфор) и остатками
большой бороздки, что связано с их доступностью.
Также наблюдается очень мало контактов с остатками малой бороздкой (ни одного полярного), это можно объяснить труднодоступностью малой бороздки.
В целом, неполярных контактов больше полярных.
Упражнение 3
С помощью программы nucplot была получена популярная схема ДНК-белковых контактов, полученные изображения вы можете видеть ниже.
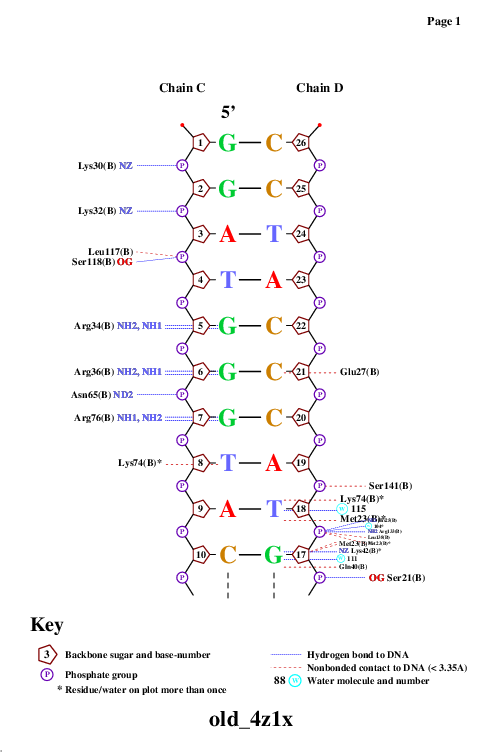 | 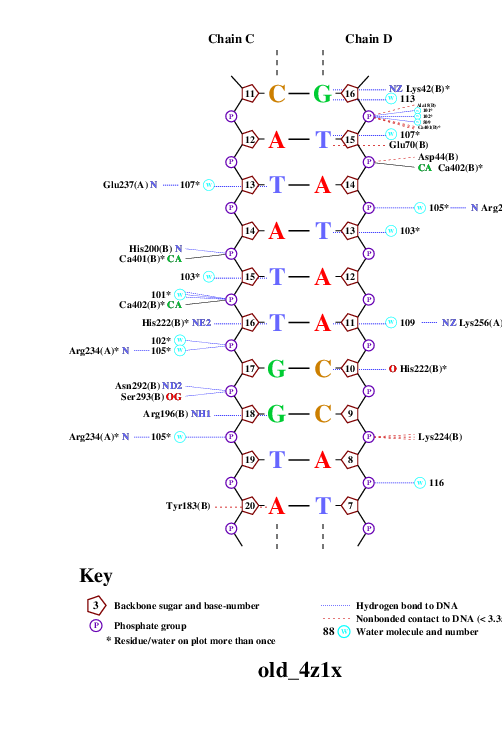 |
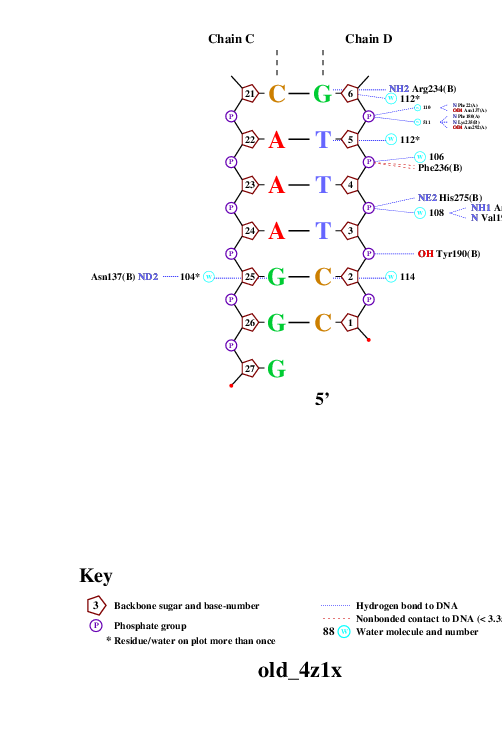 | 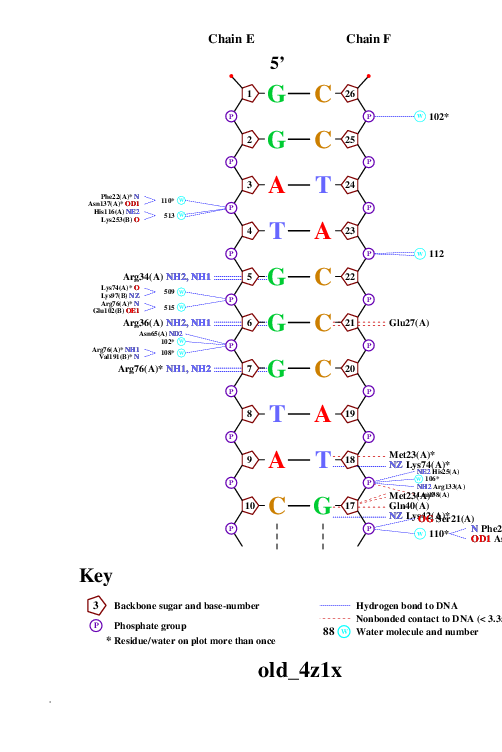 |
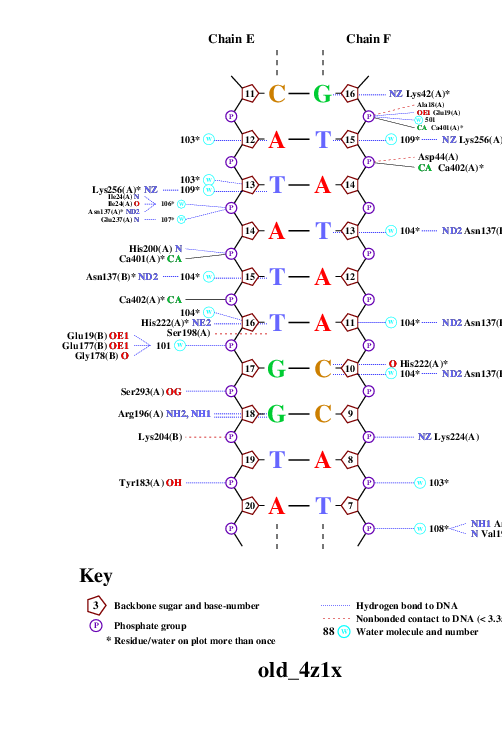 | 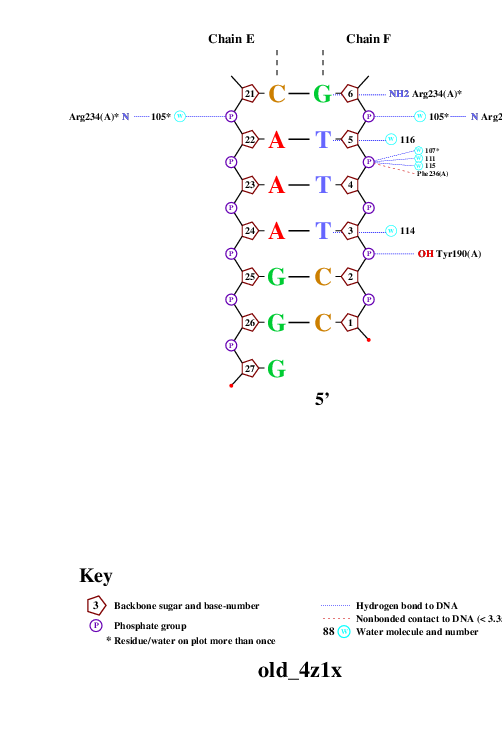 |
| Рис. 2. Популярная схема ДНК-белковых контактов. |
Упражнение 4
Аргинин - аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК;
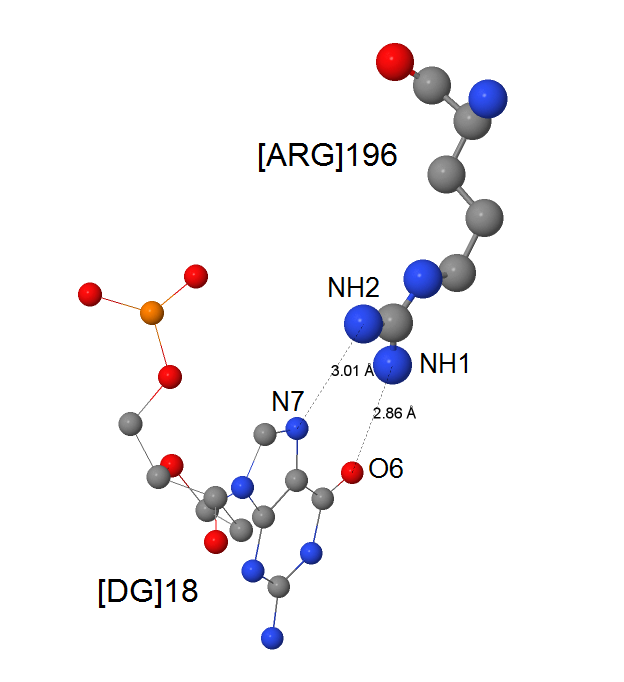 |
| Рис. 3. Изображение контакта между аргинином и гуанином. |
Аргинин и глутаминовая кислота - аминокислотные остатки, на мой взгляд, наиболее важные для распознавания последовательности ДНК, так как эти остатки способны
к специфическому связыванию с ДНК. Ниже вы можете видеть изображения, полученные с помощью JMol и иллюстрирующие контакты этих аминокислотных
остатков с ДНК.
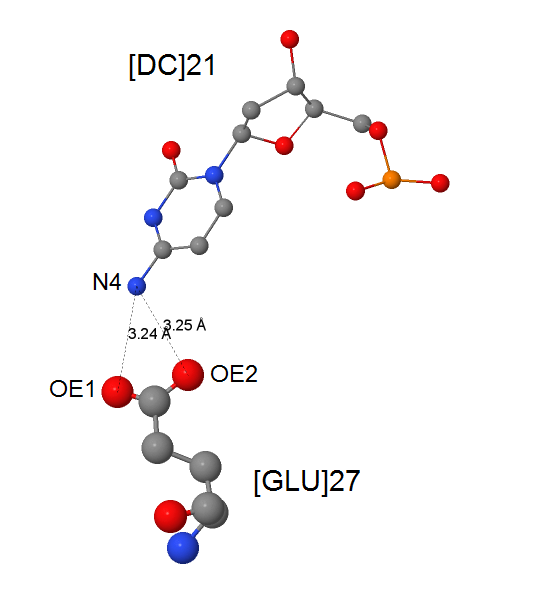 |
| Рис. 3. Изображение контакта между глутаминовой кислотой и цитозином. |
