1. Несколько файлов в формате fasta собрать в единый файл
Команда:
seqret "seq*.fasta" all.fasta
2. Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
Команда:
seqretsplit all.fasta
3. Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
Команда:
seqret @gb.txt fasta:list.fasta
4. Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле.
Команда:
transeq -table 0 list.fasta proteins.fasta
5. Транслировать данную нуклеотидную последовательность в шести рамках.
Команда:
transeq -frame 6 list.fasta proteinsf6.fasta
6. Перевести выравнивание из fasta-формата в формат .msf
Команда:
seqret align.fasta msf::align.msf
7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными.
Команда:
infoalign align.msf -refseq 2 -only -name -idcount stdout
 |
| Рис. 1. Результат выдачи |
8. (featcopy) Перевести аннотации особенностей в записи формата .gb в табличный формат .gff
Команда:
featcopy sequence.gb sequence.gff
10. Перемешать буквы в данной нуклеотидной последовательности.
Команда:
shuffleseq kamp5.fasta newgene.fasta
Далее для полученной последовательности с помощью blastn было проверено сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке
данных. Для этого был запущен blastn с порогом E = 10. В итоге находок с E-value < 0.1 не оказалось. Результат работы blastn вы можете видеть ниже.
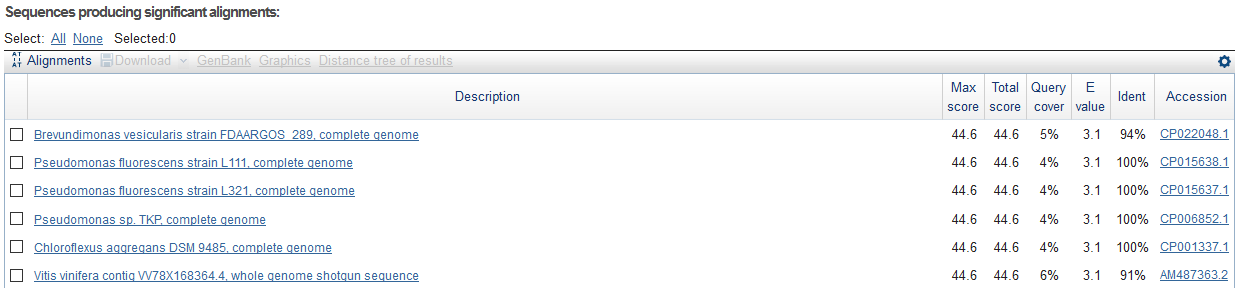 |
| Рис. 2. Результат работы blastn для случайной нуклеотидной последовательности. |
19. Создать три случайные нуклеотидные последовательности длины сто.
Команда:
makenucseq -amount 3 -length 100 mynucseq.fasta
Источники:
[1]: NCBI Nucleotide: Methanocaldococcus jannaschii DSM 2661, complete genome
