Практикум 9
Упражнения с пакетом программ EMBOSS
- (seqret) Несколько файлов в формате fasta собрать в единый файл
Исходные данные: 1.fasta и 2.fasta.
Команда:
seqret "*.fasta" both.fasta
output both.fasta
- (seqretsplit) Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
Исходные данные: both.fasta.
Команда:
seqretsplit both.fasta
output: 2973.002.fasta 2973.fasta
- (transeq) Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле
Исходные данные: both.fasta.
Команда:
transeq -table 0 both.fasta proteins.fasta
output: proteins.fasta
- (transeq)Транслировать данную нуклеотидную последовательность в шести рамках
Исходные данные: 1.fasta.
Команда:
transeq -frame 6 1.fasta prot_ws.fasta
output: prot_ws.fasta
- (seqret) Перевести выравнивание и из fasta формате в формат .msf
Исходные данные: align.fasta.
Команда:
seqret align.fasta msf::align.msf
output: align.msf
Сравнение геномов
С помощью программы blast2seq алгоритма BLASTN построена карта локального сходства двух бактерий: Helicobacter pylori Cuz20 (e-proteobacteria) в качестве Query (ось Х) и Helicobacter pylori Shi470 (e-proteobacteria) в каестве Subject (ось Y)
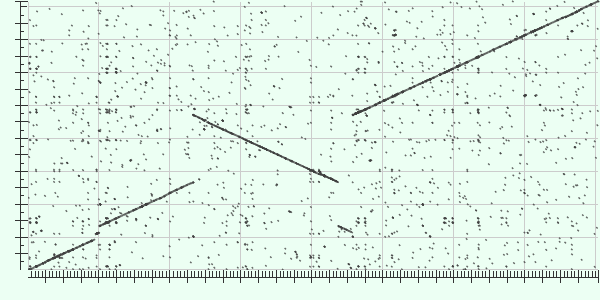
| Таблица 1. Параметры парного выравнивания геномов в blast2seq |
| Max score |
Total score |
Query cover |
E-value |
Ident |
| 2.067e+05 |
2.929e+06 |
98% |
0.0 |
98% |
Крупные эволюционные события:
Инверсия: 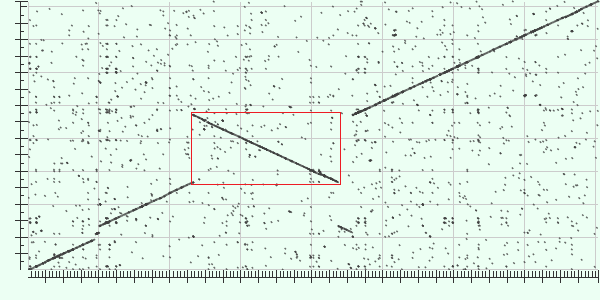
Делеция: 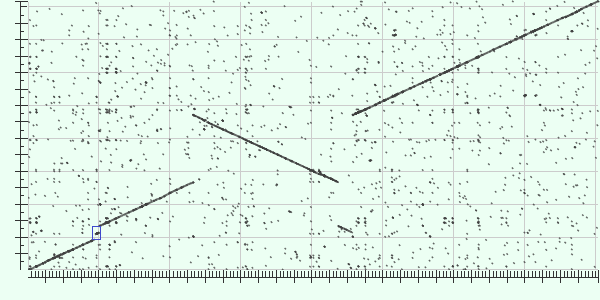
Вставка: 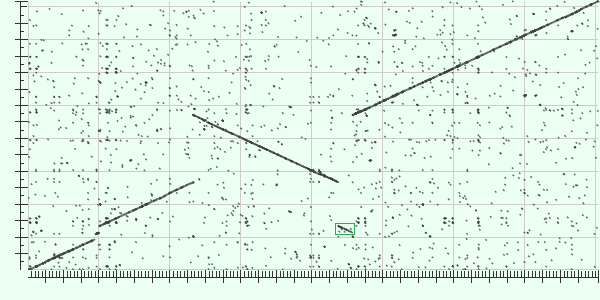
Term 3
Main page
© Artemiy Polozhintsev (Артемий Положинцев) 2016