
В данном практикуме необходимо определить сайты связывания данного транскрипционного фактора в данном участке хромосомы. Файлы .fastq с ридами Illumina, полученные в результате ChIP-seq эксперимента, разделены на отдельные файлы, соответствующие участкам хромосом.
Контроль качества с помощью Fastqc показал, что с данными можно работать, дополнительная обработка с помощью Trimmomatic не требуется.
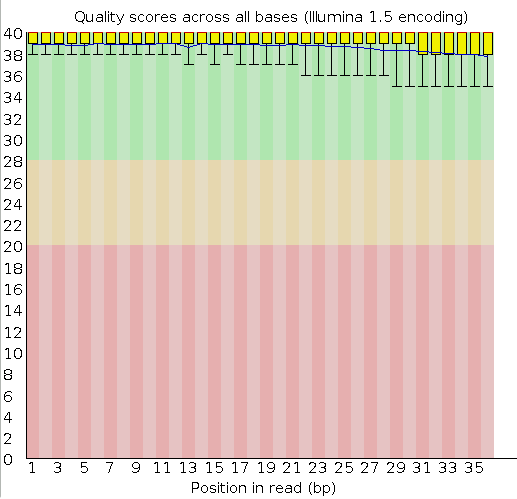
Качество позиций в ридах.
Использованные команды
| Команда | Значение |
| bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk38.fastq > chipseq_chunk38.sam | Картирование ридов из файла chipseq_chunk38.fastq, выдача записана в файл chipseq_chunk38.sam. |
| samtools view -bo chipseq_chunk38.bam chipseq_chunk38.sam | Конвертация файла в формате sam в бинарный файл bam. Опция -b - конвертировать в bam, -o - после этой опции нужно писать выходной файл. Опция -S устарела, применялась в ранних версиях, если входной файл был в формате sam. |
| samtools sort chipseq_chunk38.bam -T chip_temp -o chipseq_chunk38.sorted.bam | Сортировка bam-файла с использованием временного файла chip_temp и выходного chipseq_chunk38.sorted.bam. |
| samtools index chipseq_chunk38.sorted.bam | Индексирование сортированного файла.s |
| samtools idxstats chipseq_chunk38.sorted.bam > chipseq_chunk38.idxstats | Количество картированных и некартированных ридов по хромосомам и контигам. |
| samtools view -c chipseq_chunk38.sorted.bam | Выдаёт количество картированных ридов. |
| macs2 callpeak -t chipseq_chunk38.sorted.bam --nomodel | Получение файлов с пиками |
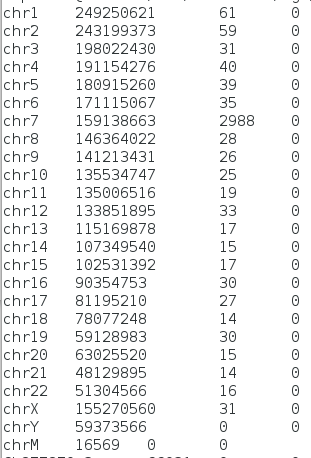
Часть файла chipseq_chunk38.idxstats с количеством ридов по хромосомам. Из этого заключаю, что этот кусок ChIP-seq принадлежал 7 хромосоме. С помощью программы MACS удалось получить два пика на 7 хромосоме. Ширина первого (слева направо) пика - 207 п.н., второго - 485 п.н. Пики лежат вне генов, скоры - 7.87 и 15.11 соответственно. В файле .narrowPeak есть ещё один пик - на контиге JH159134.2.
Визуализация файла .narrowPeak в UCSC Genome Browser. Красным помечены пики.