Анализ качеста чтений
| Название файла/программы | Команда | Результат | Описание |
| Исходные файлы | /nfs/srv/databases/ngs/azbukinanadezda/pr12/rep1 и rep2 | - | - |
| 1. Анализ качества чтений программа FastQC | установлена на компьютер, на вход получает файл .fastq | chr2.1_fastqc.html
chr2.2_fastqc.html |
Картирование чтений и анализ выравнивания
| Название файла/программы | Команда | Результат | Описание |
| 2.Картирование чтений Путь до файлов, содержащих программу Hisat2 export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Использовался индексированный файл с прошлого практикума /nfs/srv/databases/ngs/azbukinanadezda/chr2
Построение выравниваний референса и прочтений: hisat2 -x /nfs/srv/databases/ngs/azbukinanadezda/chr2 -U /nfs/srv/databases/ngs/azbukinanadezda/pr12/rep2/chr2.2.fastq --no-softclip >chr2.2.sam | /nfs/srv/databases/ngs/azbukinanadezda/pr12/chr2.2.sam или chr2.1.sam |
Команда hisat2 выравнивает прочтения и референс. Флаг -x указывает на путь к проиндексированному референсу, флаг -U - к файлу с ридами. Флаг --no-softclip запрещает подрезать нуклеотиды с концов ридов для выравнивания, флаг --no-spliced-alignment не использовался, так как мы анализируем РНК, состоящую из экзонов, а сравниваем с референсной ДНК, содержащую интроны. |
| 3. Анализ выравнивания пакет samtools |
Переведение в бинарный формат : samtools view -bS chr2.2.sam > chr2.2.bam
Сортировка по координате в референсе начала чтения: samtools sort chr2.2.bam chr2.2sorted Индексация файла samtools index chr2.2sorted.bam | /nfs/srv/databases/ngs/azbukinanadezda/pr12/chr2.2sorted.bam или chr2.1sorted.bam | samtools view переводит файл с выравниваниями в бинарный формат. samtools sort сортирует полученный бинарный файл по начальным координатам (по умолчанию). Затем файл индексируется. |
Сравнение визуализации в IGV
На рисунке 1 представлено сравнение визуализации ресеквенирования ДНК (А) и транскриптома (В). Видно, что на втором рисунке риды легли в большинстве своем на экзоны гена, а на первом рисунке на всю референсную последовательность. Также риды при секвенировании транскриптома в среднем короче по длине, чем при ресеквенировании ДНК.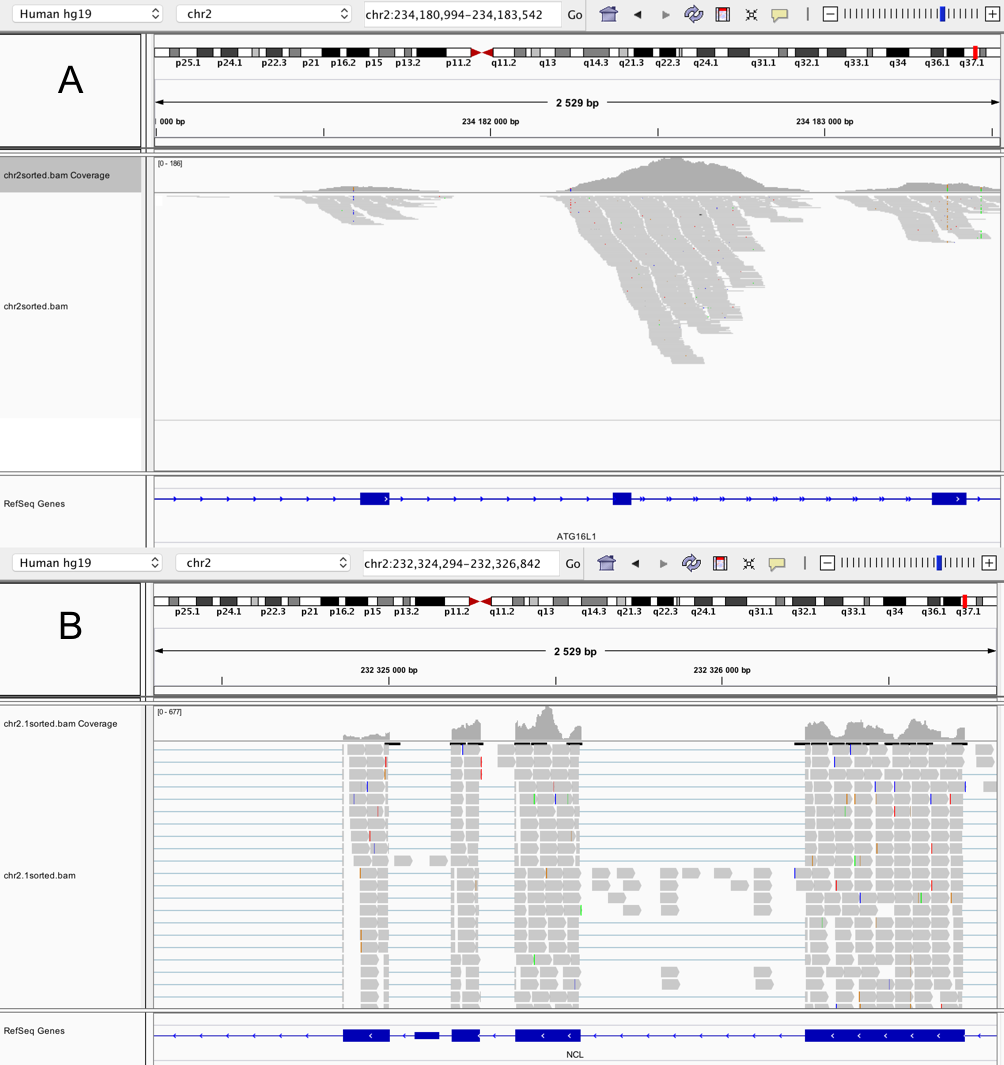
Анализ результатов с помощью bedtools
| Название файла/программы | Команда | Результат | Описание | 4. Пакет Bedtools
Путь до него:PATH=${PATH}/P/y14/term3/block4/SNP/bedtools2/bin | Перевод в формат, с которым работает bedtools: bedtools bamtobed -i chr2.1sorted.bam >chr2.1.bed | /nfs/srv/databases/ngs/azbukinanadezda/pr12/chr2.1.bed или chr2.2.bed |
| Пересечение с разметкой | bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b chr2.2.bed -c > chr2.2intersect.bed | /nfs/srv/databases/ngs/azbukinanadezda/pr12/chr2.2intersect.bed или chr2.1intersect.bed | Флаг -a указывает "референс", с которым будут сравниваться файлы, указанные под флагом -b (под этим флагом могут быть указаны несколько файлов). флаг -с считает количество покрытий участка файла под флагом -а ридами из файла -b. |
| Сортировка | Так как референсный файл содержит информацию о разметке всего генома, то выходной файл будет довольно большим. Чтобы было легче с ним манипулировать, необходимо отсортирвоать выдачу. Впринципе, у команды intersect есть опции -sorted и -sortout, однако из-за наличия в референсной разметке нестандартных записей, они не работали. Поэтому использовалась стандартная команда bash sort -k 6 -r chr2.2intersect.bed > chr2.2intersectsorted.bed | nfs/srv/databases/ngs/azbukinanadezda/pr12/chr2.2intersectsorted.bed или chr2.1intersectsorted.bed | Флаг -k указывает номер колонки, по которой происходит сортировка, поумолчанию она по возрастанию. -r переворачивает выдачу. Впринципе, можно было обойтись без него и просматривать результат не командой head, а командой tail, которая показывает последние строки файла. |
Кроме того, немного ридов легли на AC017104.2. Его координаты hg19 chr2:232,316,906-232,317,864, прямая цепь. Этот белок является неохарактеризованным.
Во второй реплике кроме описанных генов, часть ридов легло на ген TBC1D8.Координаты: hg19 chr2:101,623,690-101,767,846 размер: 144,157 п.н. , содержит 20 экзонов и 3 варианта транскрипта, расположен на обратной цепи. Этот белок может проявлять ГТФ-азную активирующую активность для белков семейства Rab (регуляторные белки). Сjдержит аргининовые и глутаминовые "пальцы", которые являются определяющими для ГТФ-азной-активирующей активности.
Больше чтения никуда не легли, то есть все оказались в пределах генов.
Если говорить о количестве покрытий этих генов, то тут возникают небольшие трудности. Число ридов в файле fastq и на выходе hisat и bamtobed 12507 и 7126 для первой и второй реплик соответственно. А на выходе команды intersect их 158 425 и 88 330. И в первой и во второй реплике число ридов увеличилось в одинаковое 12,46 раз. То есть мультипликация произошла на этапе intersect. Если учитывать все мультипликации, то в реплике один AC017104.2 покрыт 18 ридами, NCL -157459, SNORA75 -120, SNORD20 -294, SNORD82 -534, а в реплике два -AC017104.2 покрыт 6 ридами, NCL -88390, SNORA75 -90, SNORD20 -174, SNORD82 -165, TBC1D8 -5. Если в excel удалить все одинаковые строчки, то в первой реплике окажется, что AC017104.2 покрыт 9 ридами, NCL -100 691, SNORA75 -40, SNORD20 -98, SNORD82 -178, а в реплике два -AC017104.2 покрыт 3 ридами, NCL -56 824, SNORA75 -30, SNORD20 -58, SNORD82 -55, TBC1D8 -5. Как видно, это не решает проблему с мультипликациями числа ридов. Файл с расчетами здесь.