1.Предсказание вторичной структуры заданной тРНК
Вторичная структура тРНК представляет собой так называемый "клевер" и представлена на рисунке 1.
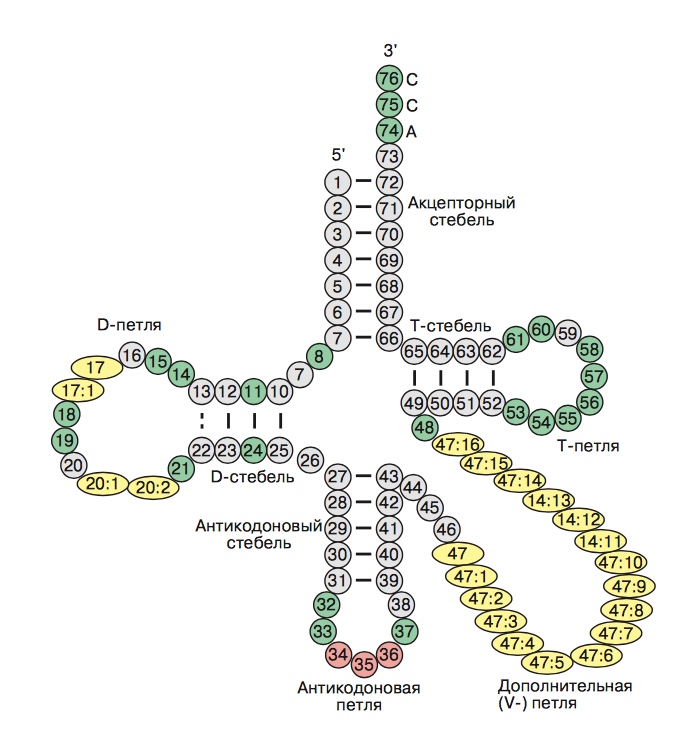
В данном задании предлагалось сравнить три различных алгоритма предсказания вторичной структуры тРНК: поиск инвертированных повторов (программа einverted из пакета EMBOSS), неспециализированная программа, определение вторичной структуры из первичной (файл 2cv0.pdb) с помощью программы find_pair и с помощью программы RNAFold, реализующей алгоритм Зукера, основанный на подсчете энергии молекулы. Как оказалось, программа einverted дает совсем неполный результат. При уменьшении штрафа за гэпы и порогового уровня появляется много "стеблей" комплементарность которых находится в районе 70%. Кроме того, предложенные вараинты совсем не соответствуют действительности. Лучший варинат, которого мне удалось достигнуть - это найти акцепторный стебель.
Score 15: 5/5 (100%) matches, 0 gaps
3 cccca 7
|||||
69 ggggt 65
Алгоритм Зунка практически правильно предсказал реальную структуру тРНК. Лучший вариант работы алгоритма представлен на рисунке 2.
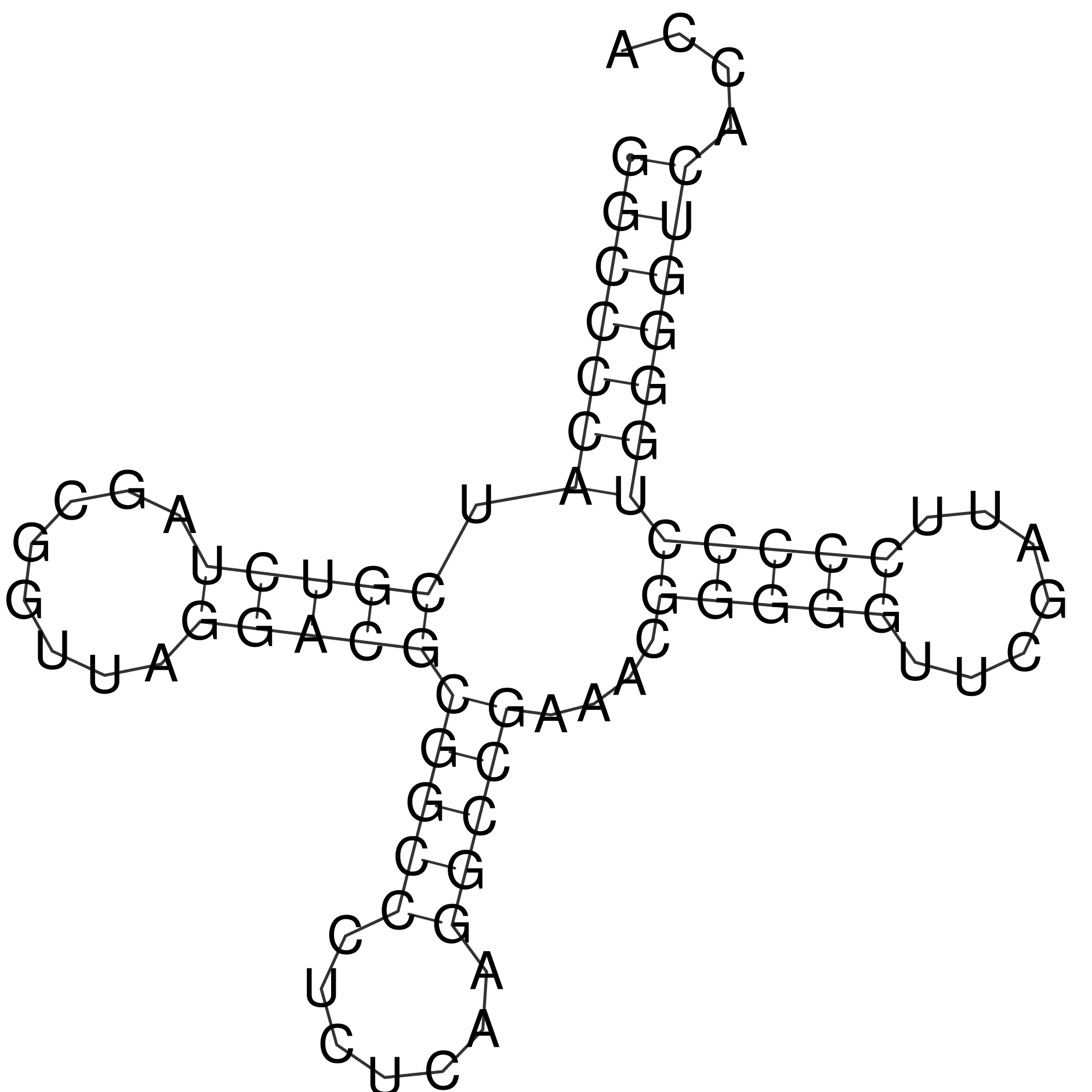
Сравнение результатов разных алгоритмов представлено в таблице 1. Как видно из таблицы, специализированная программа дает результат точнее, чем неспециализированная. Этот результат довольно близок к реальному, поулченному на основании третичной структуры, но все же есть небольшие неточности, поэтому данный алгоритм может быть использован для изучения вторичной структуры тРНК, однако необходимо помнить, что эти данные отражают лишь общий образ вторичной структуры, и никак не гарантирует абсолютной точности. (Это происходит из-за того, что такие программы используют алгоритмы, построенные на подсчете минимальной энергии. В реальности же тРНК находится не в сферическом вакууме и взаимодействует с другими молекулами, которые могут влиять на ее вторичную структуру).
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' 501_572 3'
502_571 503_570 504_569 505_568 506_567 507_:566 |
Предсказано 5 пар из 7 реальных | Предсказано все 7 |
| D-стебель | 5' 510_525 3'
511_524 512_523 513_522 514_521 |
Не предсказано | Предсказано все 5 пар |
| T-стебель | 5' 549_565 3'
550_564 551_563 552_562 553_561 554_558 |
Не предсказано | Предсказано 5 пар из 6 |
| Антикодоновый стебель | 5' 538_532 3'
539_531 540_530 541_529 542_528 543_527 544_526 |
Не предсказано | Предсказано 5 пар из 7 |
| Общее число канонических пар нуклеотидов | 21 | 5 | 18 |
2. Поиск ДНК-белковых контактов в заданной структуре
| task 1 | Текст скрипта |
| task 2 | Текст скрипта |
| Resume | |
| Для переключения изображений нажмите "Resume" | |
На представленном выше апплете вы можете ознакомиться с ДНК-белковым комплексом 1dfm.pdb. При нажатии на первую клавишу вы увидите выделенными шариками различные группы атомов. При нажатии на вторую клавишу-различные полярные и неполярные взаимодействия между ДНК и белком. Для переключения между изображениями нажмите 'Resume'. Под полярными атомами будем понимать атомы кислорода и азота, а под неполярными - углерода, фосфора и серы. Для полярных контактов наибольшим расстоянием будет 3,5 Å, для неполярных - 4,5. Общие сведения представлены в таблице 2. Количество связей считалось как наименьшее количество атомов, выделяемыми атомами белка или днк, находящихся на меньших, чем указанных выше растояниях от другого биополимера. Как видно из полученной таблицы, больше всех неполярных связей образуется с атомами дезоксирибозы, больше всех полярных с кислородами фосфорного остатка. Больше всех связей с белком образует пентоза. Отсюда можно сделать вывод, что белок при связывании в комплекс с ДНК образует связи с сахарофосфатным остовом, причем неполярные. То есть взаимодействие реализуется за счет гидрофобного эффекта. Эти данные подтверждают полученные мною выводы из 2 практикума прошлого семестра. Интересно заметить, что атомы малой бороздки ДНК вообще не образуют связей с белком.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 7 | 43 | 50 |
| остатками фосфорной кислоты | 20 | 13 | 33 |
| остатками азотистых оснований со стороны большой бороздки | 6 | 10 | 16 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 0 | 0 |
Затем с помощью программы nuclprot была получена популярная схема ДНК-белковых контактов. Как я поняла, основным критерием контакта для программы служит расстояние 3,5 Å. Поэтому, чтобы получить информацию, касающуюся только ДНК-белковых контактов из исходного файла необходимо было удалить молекулы воды. На выход программа выдает несколько файлов, содержащих информацию о полярных, неполярных, коваленьных и всех вместе связах, а также иллюстративный файл формата ps. Полученный результат можно увидеть на рисунке 3. Скачать файл, содержащий информацию о связях здесь.
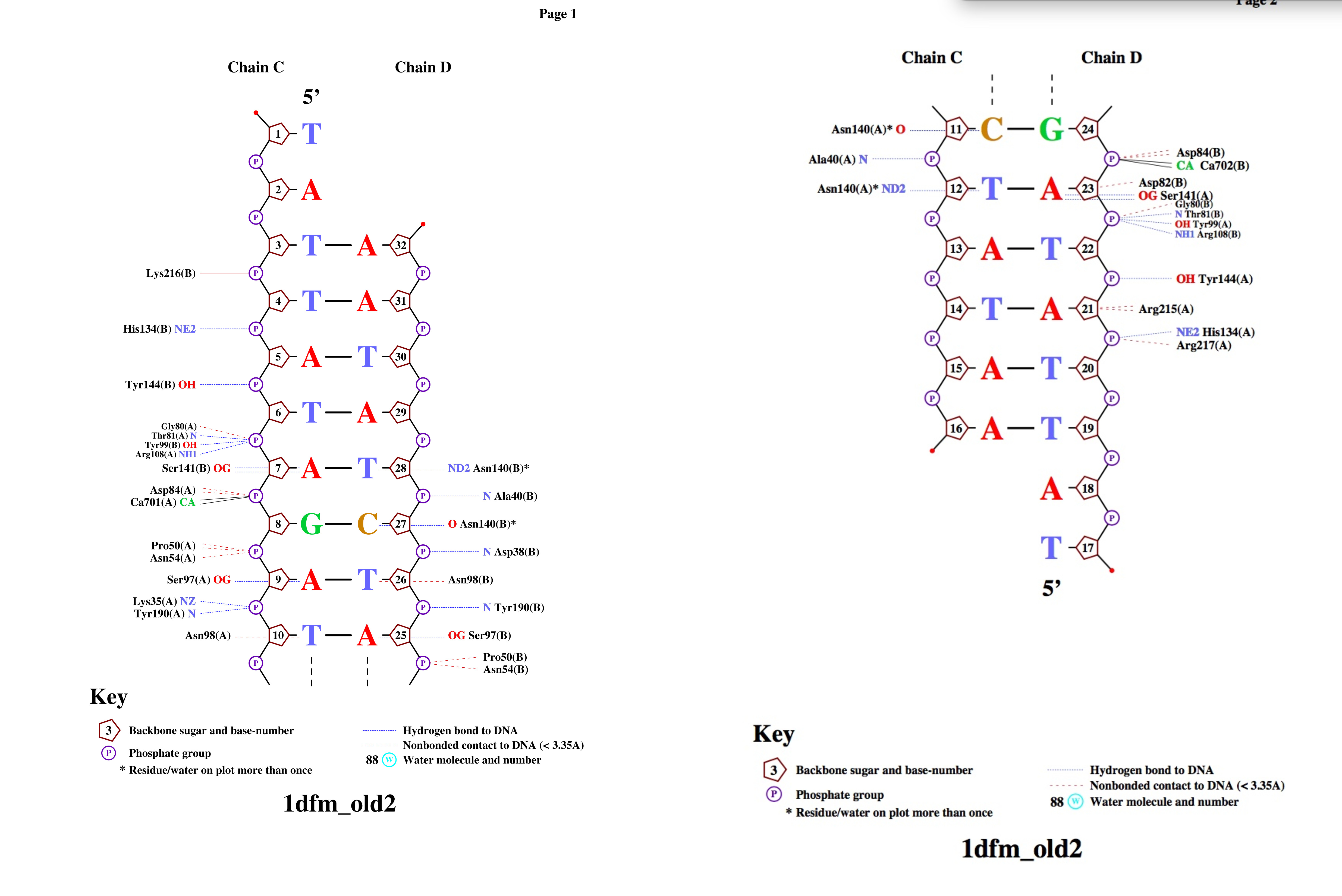
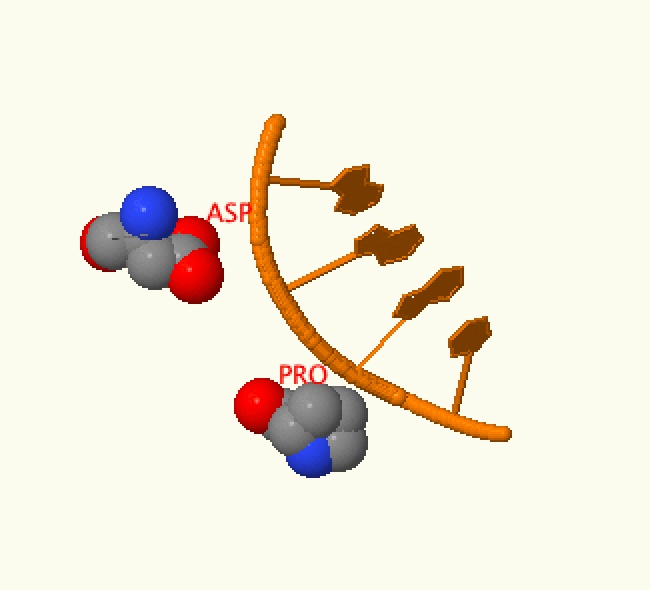
Проанализировав файл, полученный на выходе программы nuclprot, формата .bond, можно найти информацию о количестве связей аминокислот с ДНК. Чтобы облегчить себе задачу, я удалила из файла pdb молекулы воды и запустила заново nuclprot (чтобы получить информацию только о ДНК-белковых взаимодействиях). Аминокислотой, связанной с наибольшим числом атомов ДНК оказалась Asp 84, связанная двумя водородными связями с OH-группой фосфата 8. Также 2 водородные связи с OH-группой фосфата 9 связана аминокислота Pro 50 (так как структура белка симметрична, эти аминокислоты расположены и на А и на В цепях белка). Изображение взаимодействия представлено на рисунке 4. Так как это два соседних фосфата, а аминокислоты не соседние и оказываются позиционирвоанными рядом из-за третичной структуры белка, и образуют максимальное (по 2 на аминокислоту) число связей, я считаю, что они являются важными аминоксилотами, обеспечивающими связь ДНК-белок.
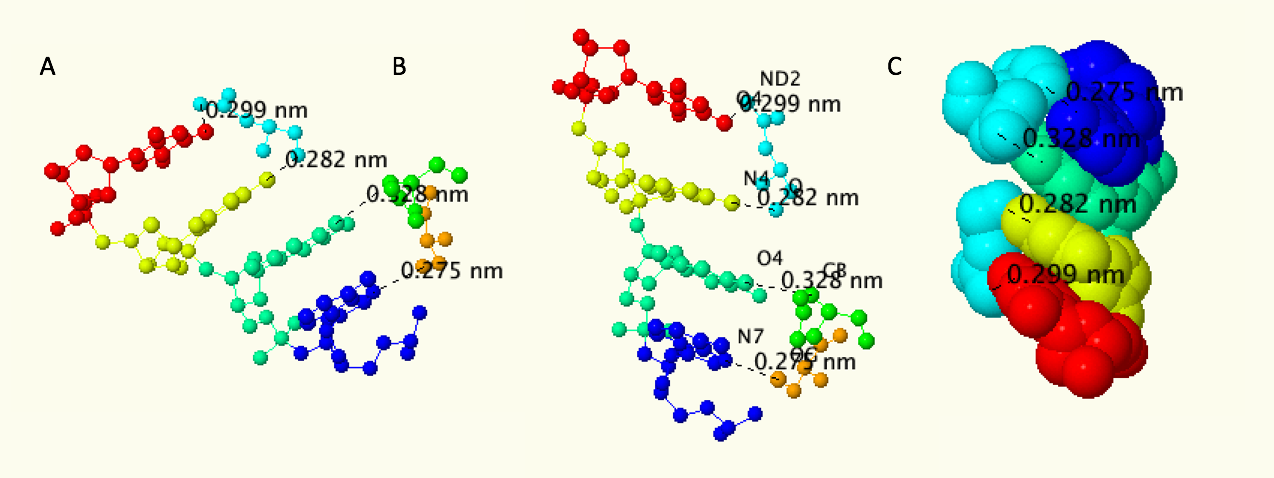
Говоря о специфичности узнавания последовательности ДНК, нужно обратиться к схеме, представленной на рисунке 3. Можно увидеть, что между цепью С (9-12 нуклеотиды) и белковой цепью А , а также D (23-26 нуклеотиды) и белковой цепью B есть связи между атомами нуклеотидов большой бороздки и аминокислотными остатками. На рисунке 6 можно увидеть визуализацию связей между цепями A и C. ASN 140 образует целые две связи: одну с Тимином 12, другую - с Цитозином 11, SER 97 - с Аденином 9, ASN 98 -с Тимином 10 (Полный список связей: ([ASN]140:A.ND2 or [DT]12:C.O4 or [DC]11:C.N4 or [ASN]140:A.O or [SER]97:A.OG or [DA]9:C.N7 or [ASN]98:A.CB or [DT]10:C.O4)). Так как эти нуклеотиды, расположенные по порядку, образуют связи с белком, можно предположить, что именно этот мотив ДНК и узнается.
3. Список литературы
[1] http://kodomo.fbb.msu.ru/FBB/year_07/term3/tRNA.pdf