1.Чтение последовательностей ДНК, полученных из капиллярного секвенатора
Мне было предложено два файла для анализа, прямая и обратная последовательности. Скачать их можно по ссылке в формате .ab1. Для анализа полученных хроматограмм использовалась программа 4Peaks [1]. Я открыла в двух окнах эти файлы, в файле с обратной последовательностью перешла к комплементарной цепочке.
В файле с прямой последовательностью я обрезала первые 60 нуклеотидов, так как на этом фрагменте совершенно невозможно судить о последовательности: шум сливается с с сигналом, пики неравномерные, без четких зубцов (См. рисунок 3, верхняя часть). Конечную часть этого файла я обрезать не стала, на мой взгляд, по ней можно прочитать последовательность нукеотидов. С 60 по 130 нуклеотид еще довольно сложно разобрать принадлежность нуклеотидов, много неоднозначностей и перекрываний, однако со 130 все вполне прилично, только последние 10 нуклеотидов имеют неровные большие пики с размытыми зубцами. В общем я думаю, что это достаточно хорошая хроматограмма, с низким урвонем шума по все хроматограмме (примерно 10% от нормального сигнала). В случае файла с обратной последовательностью я буду употреблять нумерацию для комплементарной цепочке (перевернутой). В этом файле я исключила последние 13 нуклеотидов, все по той же причине. Для этого файла последние 13 на самом деле при секвенировании были первыми, поэтому этот фрагмент содержит много шума, так как при секвенировании капиллярным секвенатором расшифровка последовательности начального фрагмента затруднена. В этом файле только первые 10 и последние 20 нуклеотидов сложно прочитать, остальные - очень ровные пики. Однако в этой хроматограмме высота самих основных пиков больше и больше уровень шума (до 20%), чем в предыдущем файле. Несмотря на это, эту хрматограмму я тоже назвала бы хорошей и "читаемой", так как неприемлимый урвоень шума был всего в 30 нуклеотидах из 600.
При выравнивании хроматограмм 90-ый нуклеотид обратной последовательности соответствовал 61-ому нуклеотиду прямой последовательности. Затем я вручную проверяла правильность соответствия нуклеотида пику, так как программа может ошибаться.
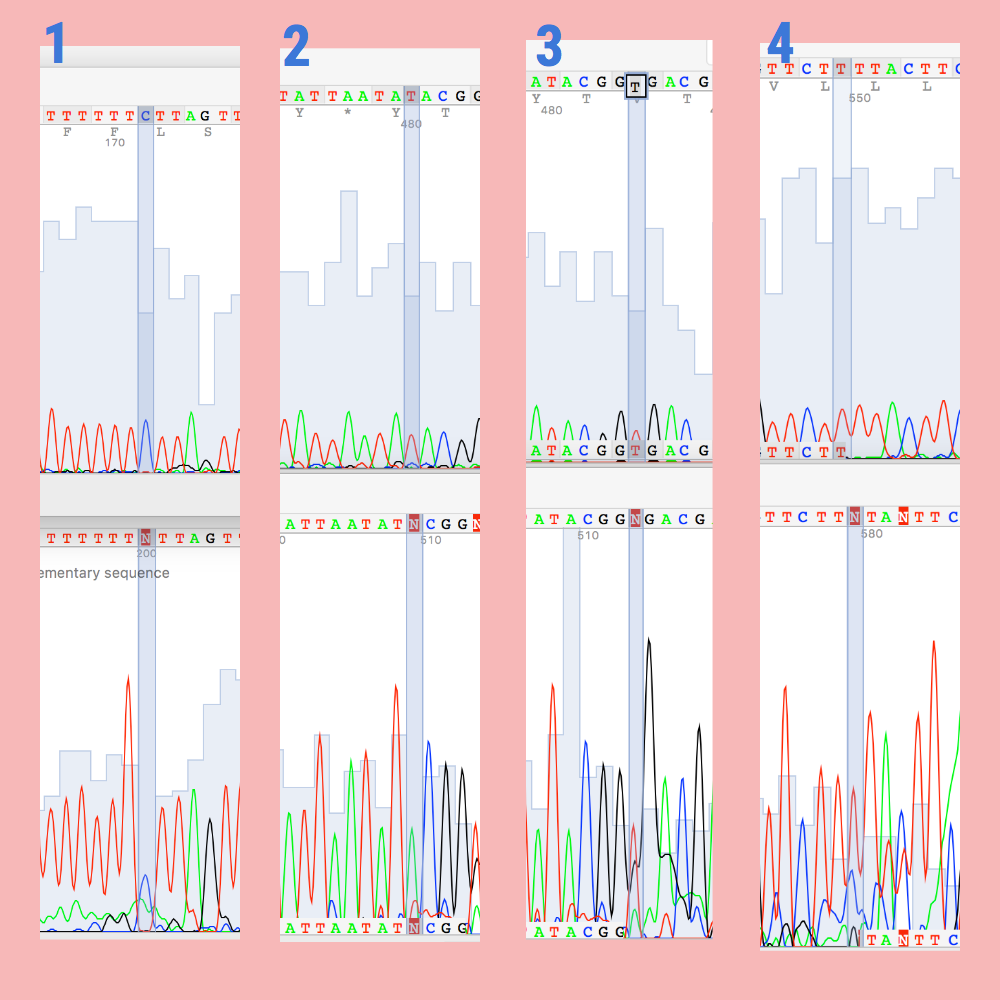
Ниже представлены 4 примера исправления нуклеотидов.
1) 200-ый нуклеотид обратной цепочки позиционировался программой как N, то есть любой из 4 возможных (подробные обозначения см) из-за шума "зеленого" флуорофора (А). Если сравнить с прямой цепочкой, там четко видно, что этот цитозин.
2) 509-ый нуклеотид обратной цепочки позиционировался как N, опять же из-за шума. Сравнив с прямой цепочкой я исправила на аденин, так как зубец "красного" флуорофора я бы не расценивала как полиморфизм, тк он очень нечеткий и не выглядит как достоверный пик нуклеотида.
3) 513-ый нуклеотид обратной цепочки позиционировался как N из-за того, что вокруг этого нуклеотида несколько гуанинов, их пики дают большой шум на тимин между ними. При сравнении с прямой цепочкой исправлено на тимин.
4) 579-ый нуклеотид обратной цепочки позиционировался как N из-за шумовой флуоресценции. Около соседних пиков также есть этот шум, однако я не расцениваю его как полиморфизм, так как зубцы не совпадают с основными, как будто это просто кусочек другой ДНК. При сравнении с прямой цепочкой исправила на тимин.
После редактирования хроматограмм, файлы были экспортированы в формате .fasta и с помощью программы needle пакета emboss были глобально выровнены (needle L39_COI_F.fasta L39_COI_R.fasta -outfile 1 -aformat3 fasta, остальные параметры, такие как штрафы за гэпы, были выбраны по умолчанию). Результат выравнивания:
# Aligned_sequences: 2 # 1: L39_COI_F # 2: L39_COI_R # Matrix: EDNAFULL # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 745 # Identity: 521/745 (69.9%) # Similarity: 526/745 (70.6%) # Gaps: 208/745 (27.9%) # Score: 2566.0


По результам выравнивания я составила варинат прочитанной последовательности ДНК. Скачать файл в формате .fasta по ссылке. Затем я убрала начальные и проблемные участки, в которых нельзя однозначно интерпретировать, какой нулеотид стоит на этом месте. Результат доступен по ссылке. Проблемные нуклеотиды обозначены строчными буквами. Затем мне стало интересно, насколько правильным получился мой результат. С помощью программы Blast nucleotides я попыталась найти похожие последовательности. Лучшим вариантом стал ген Lacuna vincta voucher 11BFMOL-0096 cytochrome oxidase subunit 1 (COI) , Query cover 88%, Identity 99%. Ссылка на этот ген. Я считаю полученный результат довольно хорошим, так как получилось практически правильно восстановить исходную последовательность гена цитохром оксидазы (1 субъединица) брюхоногого моллюска.
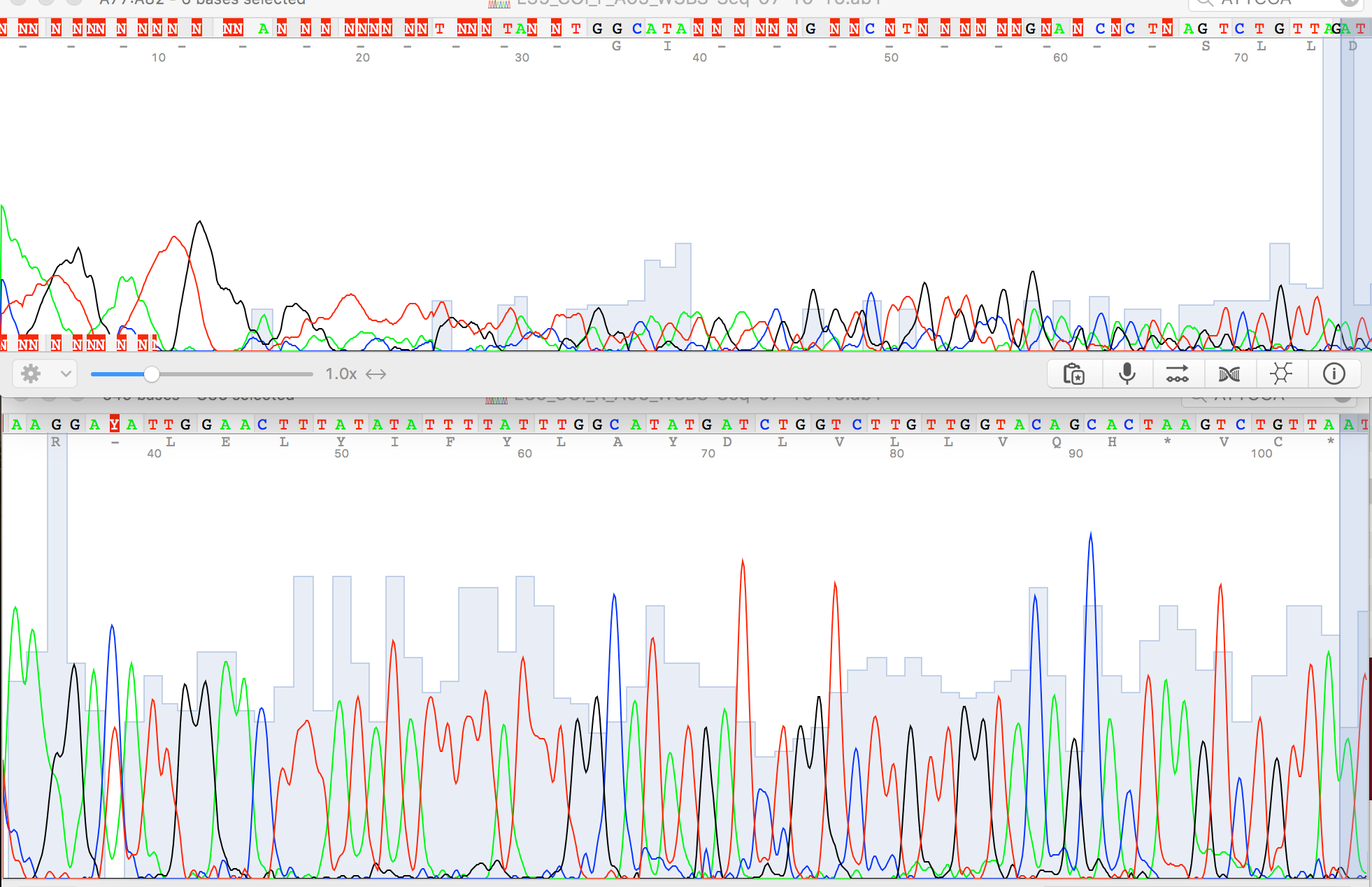
2. Пример нечитаемого фрагмента хроматограммы
3. Список литературы
[1] http://nucleobytes.com/4peaks/