Поиск ДНК-белковых контактов в заданной структуре
В программе JMol с помощью команды define можно задавать множества атомов. Определим множества следующих атомов:
- множество атомов кислорода 2'-дезоксирибозы (set1);
- множество атомов кислорода в остатке фосфорной кислоты (set2);
- множество атомов азота в азотистых основаниях(set3);
Создадим скрипт , вызов которого даёт в программе JMol последовательно следующие изображения:
- вся структура
- только ДНК (в проволочной модели);
- проволочная модель ДНК с выделенными шариками множества атомов set1,
- проволочная модель ДНК с выделенными шариками множества атомов set2,
- проволочная модель ДНК с выделенными шариками множества атомов set3.
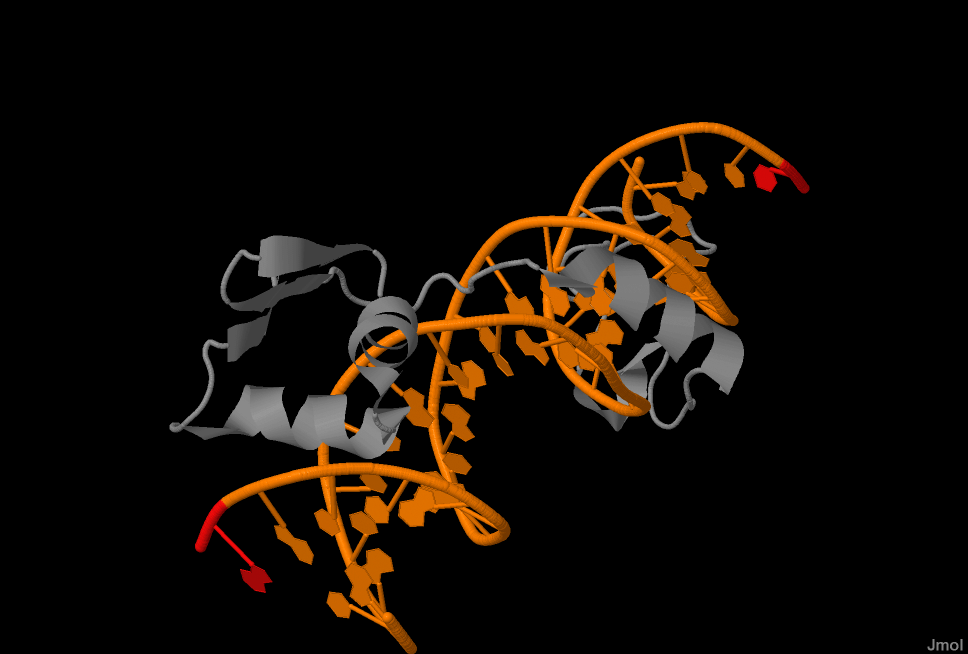 |
Рис. 1 Вся структура |
 |
Рис. 2 ДНК в проволочной модели (с помощью функции wireframe) |
 |
Рис. 3 Атомы кислорода 2'-дезоксирибозы (получены с помощью cpk, окрашены в красный цвет) |
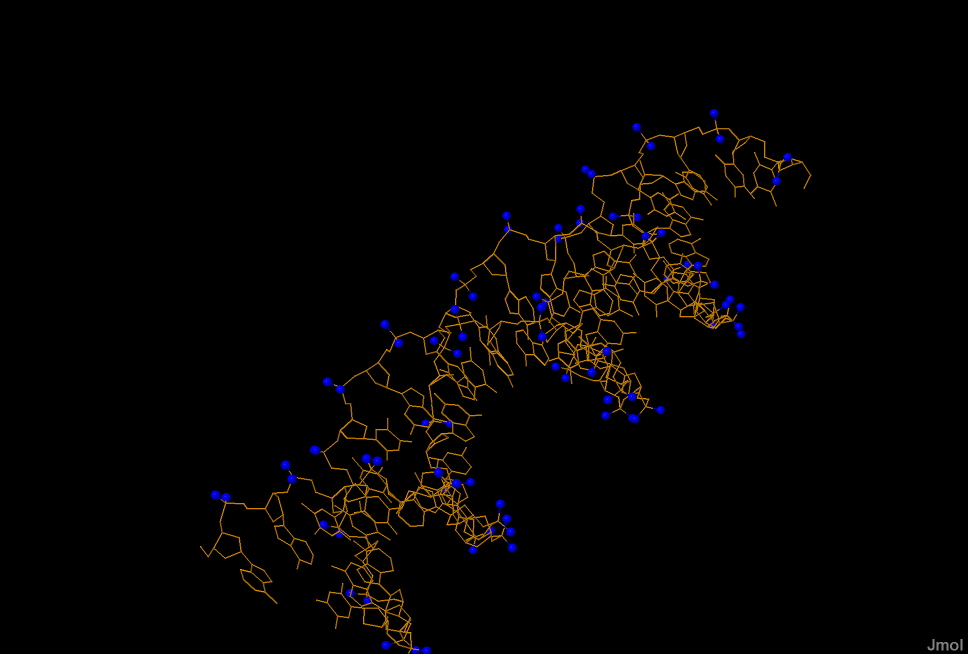 |
Рис. 4 Атомы кислорода в остатке фосфорной кислоты (получены с помощью cpk, окрашены в синий цвет) |
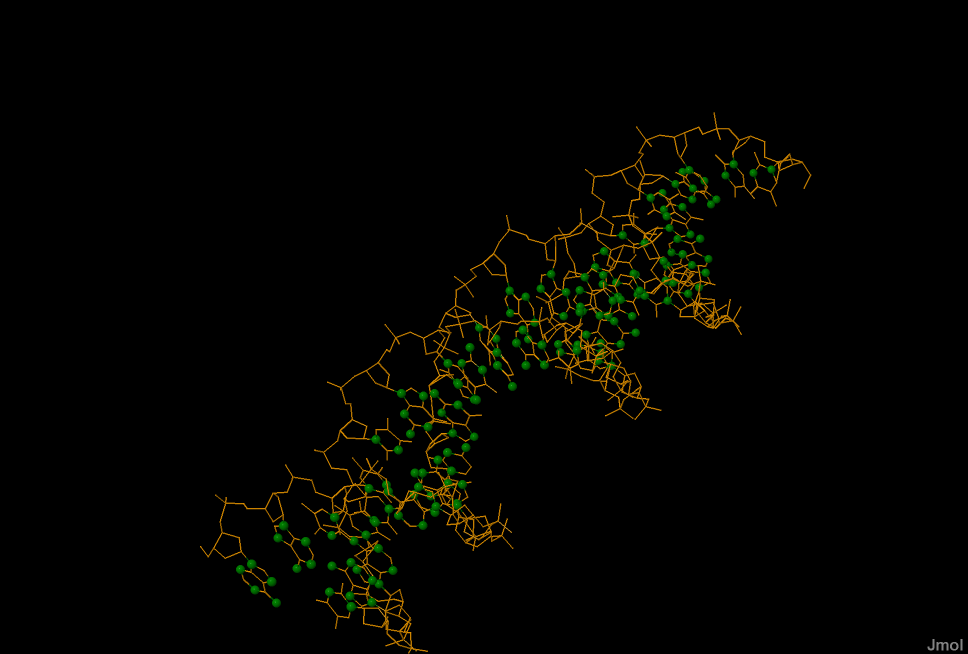 |
Рис. 5 Атомы азота в азотистых основаниях (получены с помощью cpk, окрашены в зеленый цвет) |
Поиск ДНК-белковых контактов в заданной структуре
Для того, чтобы найти межмолекулярные взаимодействия ДНК с белком, напишем скрипт.Полярными будем считать атомы кислорода и азота(расстояние не более 3,5 ангстрем), а неполярными- атомы углерода,фосфора и серы (расстояние не более 4,5 ангстрем). Результат приведен в таблице:
| контакты атомов белка с | полярные | неполярные | всего |
| остатками 2'-дезоксирибозы | 17 | 93 | 110 |
| остатками фосфорной кислоты | 21 | 80 | 101 |
| остатками азотистых оснований со стороны большой бороздки | 1 | 5 | 6 |
| остатками азотистых оснований со стороны малой бороздки | 9 | 25 | 34 |
Использование программы nucplot
C помощью программы nucplot получила схему контактов белка с ДНК. Она представлена на рис. 6.
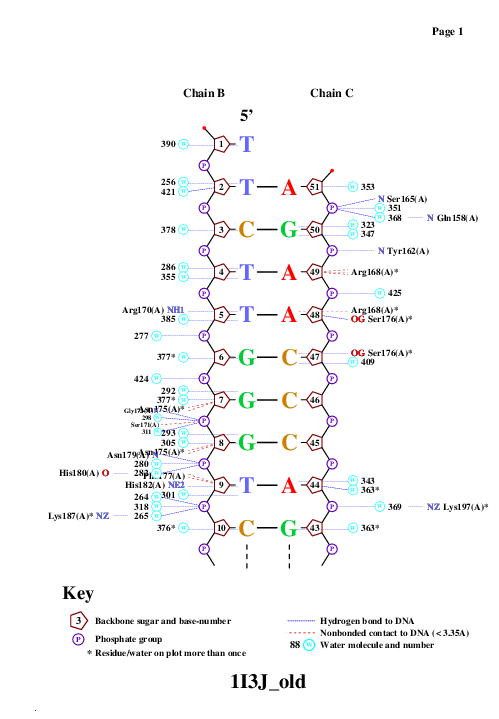 |
Рис. 6 Иллюстрация контактов цепей ДНК, полученная с помощью программы nucplot |
Максимальное количество контактов ДНК имеет с Arg168(2 контакта).Они представлены на изображении 7 полученным с помощью программы JMol:
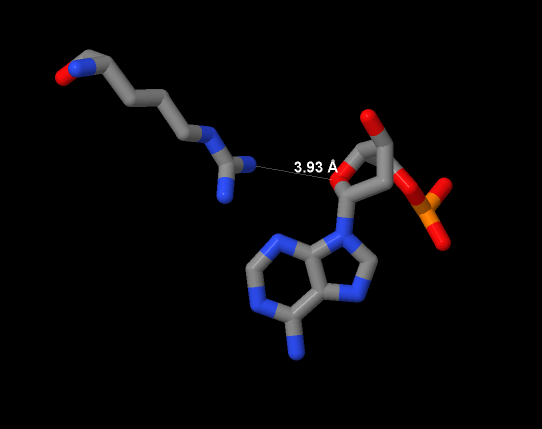 |
Рис. 7 Иллюстрация контакта Arg168 с 49A цепи С |
Если говорить о том, какой остаток важен для распознавания фрагмента ДНК, то можем отметить остатки T5 и Т9, так как в них происходит непосредственное образование водородных связей между остатком азотистого основания и боковым радикалом цепи белка.Схемы, иллюстрирующие взаимодействия приведены на рисунках 7, 8
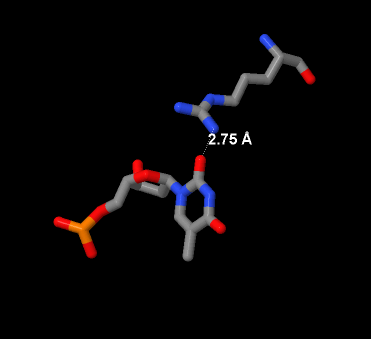 |
Рис. 8 Иллюстрация контакта Arg170 с 5T цепи B |
 |
Рис. 9 Иллюстрация контакта His182 с 9T цепи B |
Предсказание вторичной структуры тРНК
Предсказание вторичной структуры тРНК путем поиска инвертированных повторов
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях.При этом были заданы следующие параметры:
В итоге был получен файл:sequence.inv, содержащий два очень коротких выравнивания участков инвертированных последовательностей. При использовании значений "Minimum score threshold" ниже 9 в результирующем файле ничего не меняется.
Предсказание вторичной структуры тРНК по алгоритму Зукера
Программа mfold из пакета EMBOSS реализует алгоритм Зукера. Побробуем провести предсказание с отклонением энергии на 15%,в итоге получаем 10 возможных структур, однако все из них не совпадали с реальной структурой тРНК. Поэтому решили провести поиск при 5 % и при таком анализе получили 2 возможные структуры ( картинки представлены ниже). Далее попробуем провести поиск с нулевым отклонением, т.е наиболее точное предсказание( см. рис. 11). Работа проводилась в браузерной программе. Несмотря на то, что обе полученные структуры наиболее близки по энергии к оптимальной, верхняя структура более правильна, т.к сохраняет стандартную форму тРНК.
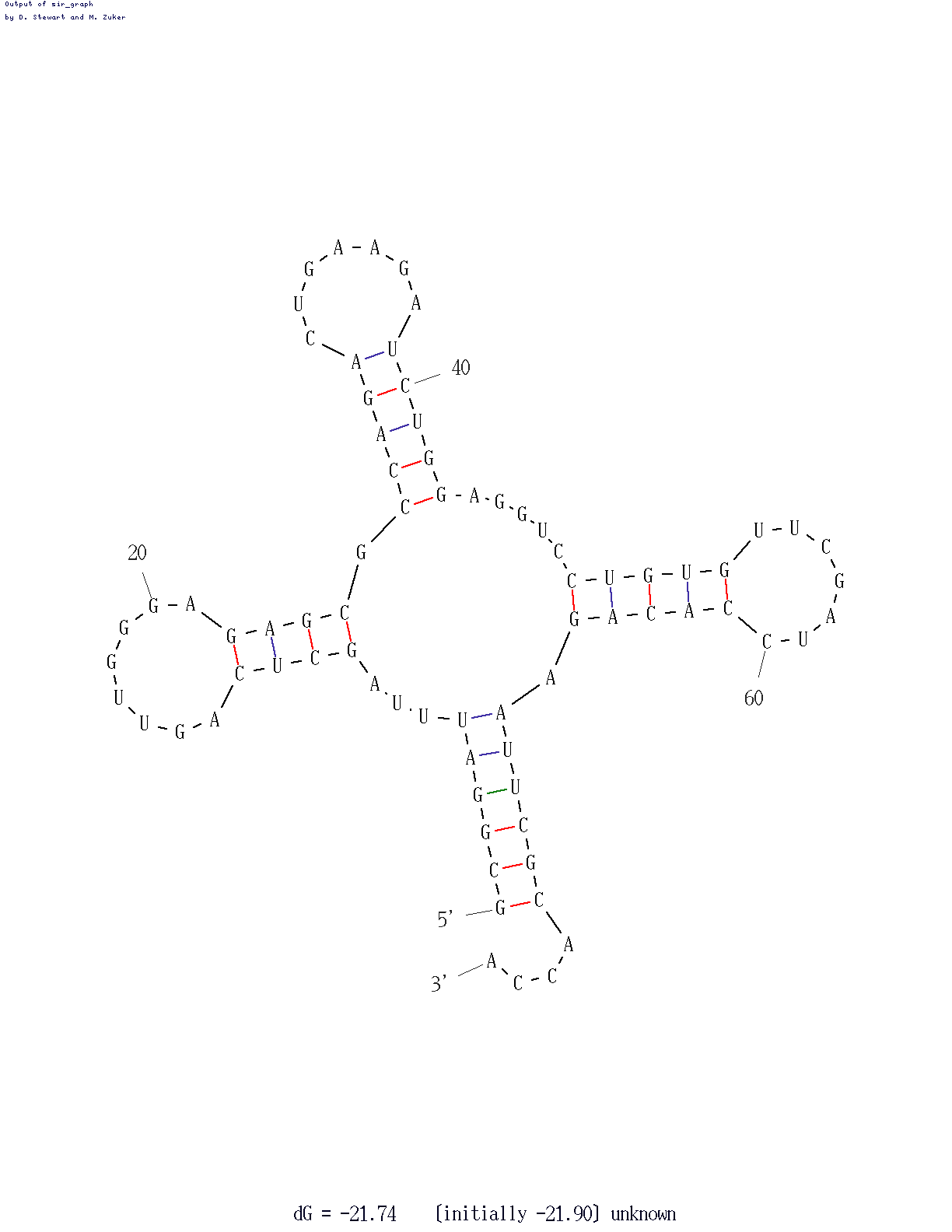 |
Рис. 10 Результат предсказания вторичной структуры тРНК с использованием алгоритма Зукера |
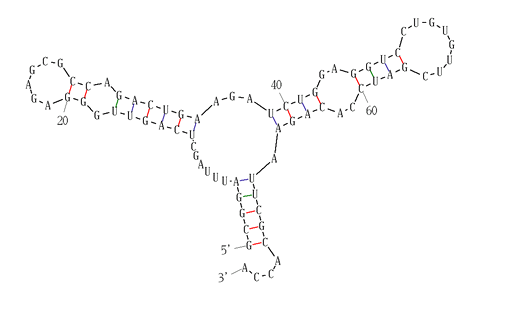 |
Рис. 11. Поиск с нулевым отклонением |
Сравнение результатов программ, с помощью которых в этом задании мы пытались предсказать вторичную структуру тРНК 1ML5 можно посмотреть в таблице:
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5'-66-71-3' 5'-2-7-3' 6 пар |
0 правильных пар | 5'-67-72-3' 5'-1-6-3' 5 правильных пар |
| D- стебель | 5'-10-13-3' 5'-22-25-3' 4 пары |
0 правильных пар | 5'-10-13-3' 5'-22-25-3' 4 правильные пары |
| Т-стебель | 5'-49-53-3' 5'-61-65-3' 5 пар |
5'-49-53-3' 5'-61-65-3' 5 правильных пар |
5'-49-53-3' 5'-61-65-3' 5 правильных пар |
| Антикодоновый стебель | 5'-26-32-3' 5'-38-44-3' 7 пар |
5'-22-31-3' 5'-39-48-3' 6 правильных пар |
5'-27-31-3' 5'-39-43-3' 5 правильных пар |
| Общее число канонических пар нуклеотидов | 22 | 15 | 20 |