Обзор генома бактерии Photobacterium gaetbulicola Gung47
Резюме
В работе охарактеризован геном Photobacterium gaetbulicola Gunng47, морской бактерии из семейства Vibrionaceae. Акцент сделан на тех характеристиках генома, которые могут указывать на особенности репликации обсуждаемой бактерии.
1. Введение
Photobacterium gaetbulicola — грамотрицательная факультативно анаэробная бактерия из рода Photobacterium (Gammaproteobacteria; Vibrionales; Vibrionaceae). Типовой штамм P. gaetbulicola Gung47 выделен из литорали на западном берегу Кореи [1]. Оптимальные условия роста для этого вида бактерий — 30 °C, pH = 7–9, 2–5 % (м/о) NaCl. Для этого вида определен состав жирных кислот, с помощью стандартных тестов охарактеризован фенотип, найдены кластеры генов, отвечающие за синтез некоторых метаболитов [1,2].
P. gaetbulicola имеет две неравные по размеру хромосомы [3]. Это характерная особенность всего семейства Vibrionaceae; считается, что бо́льшая хромосома консервативнее и несет необходимые для жизнедеятельности гены, а меньшая — более специализированные, хотя это лучше изучено для рода Vibrio, чем для других родов семейства [4]. В отличие от многих других Photobacterium, P. gaetbulicola Gung47 не имеет плазмид [3].
Также для Vibrionaceae характерно большее количество копий генов тРНК и рРНК, чем для других бактерий; это связывают с очень высокой скоростью репликации (по крайней мере, для рода Vibrio) [4]. Photobacterium profundum на момент секвенирования имел наибольшее из всех бактерий количество оперонов рРНК в геноме — 14 на большей хромосоме и 1 на меньшей [5].
В этой работе я привел основные характеристики генома P. gaetbulicola Gung47, постаравшись уделить больше внимания тем из них, которые могут быть полезны при изучении особенностей репликации этой бактерии.
2. Материалы и методы
Для работы я использовал файл с последовательностью всего генома и хромосомную таблицу (feature table), скачанные с FTP-сервера NCBI [6] (сопроводительные материалы S10 и S11). Для обработки и визуализации данных пользовался R 4.2.1, Python 3.10.4, Bash 5.10.16.
Для подсчета суммарной длины кодирующих последовательностей использовал сценарий S1. Кодирующими последовательностями здесь считаются последовательности, отмеченные в хромосомной таблице как «CDS with protein».
Для расчета cumulative GC и AT-skew использовал сценарий S2. Значение cumulative GC-skew в координате n последовательности вычисляется по формуле
\[\frac{G_n - C_n}{G_N + C_N},\]
где Gn и Сn — количества соответствующих нуклеотидов в последовательности до координаты n включительно, а GN и СN — суммарное количество G и C во всей последовательности (иначе говоря, это cumulative GC-skew, рассчитанный с использованием окна и шага размера 1, и нормированный на общее количество G и C). Для AT-skew формула аналогичная.
Для расчета GC- и AT-skew отдельно в кодирующих и некодирующих последовательностях использовал сценарий S3. Здесь значение skew нормируется не на количество G + C (A + T) во всей хромосоме, а на количество данных нуклеотидов в соответственно кодирующих или некодирующих последовательностях.
Для подсчета асимметрии распределения генов по цепям хромосомы использовал сценарий S4. Значение рассчитывается по формуле, аналогичной формуле для GC-skew, где вместо G подставляется количество генов на «+» цепи, а вместо C — на «−» цепи. За координату гена принята координата первого нуклеотида в его старт-кодоне.
Для визуализации skew использовал сценарии S5 и S6.
Для подсчета расстояния от генов до ориджина репликации использовал сценарий S7. Этот же сценарий рассчитывает значение критерия χ2 для проверки принадлежности расстояний от генов до ориджина репликации к равномерному распределению. Для вычисления значения критерия я использовал 10 карманов равной длины, т.к. в минимальной выборке, для которой был проведен тест, 60 наблюдений (гены рибосомальных белков на хромосоме 2), а карманы рекомендовано подбирать так, чтобы ожидаемое количество наблюдений в них было не меньше десяти [7]. Таблицу пороговых значений критерия взял на сайте «Медицинская статистика» [8].
Биномиальный тест проведен в R функцией binom.test() без дополнительных настроек.
В статье Hu и др. [9] на выборке из 170 видов бактерий показана корреляция между максимальной скоростью удвоения, характерной для бактерии, и такими характеристиками ее генома, как отношение количества генов тРНК к количеству генов рРНК, среднее расстояние между ориджином репликации и генами тРНК и рРНК, разница между этими расстояниями. В статье бактерии делятся на две группы — «быстрорастущие» и «медленнорастущие» — и линии тренда приводятся отдельно для этих групп, для некоторых характеристик — в логарифмических осях (см. Fig. 3 в [9]). Сценарий S12 строит линейную регрессионную модель для предсказания времени удвоения по каждой из обсуждаемых характеристик в тех же осях, в каких это сделано в статье Hu и др., но без деления бактерий на две группы. Данные из цитируемой статьи приложены как файл S13. Для построения регрессии используется функция R lm(). В полученные уравнения подставляются значения для P. gaetbulicola (расстояния до ориджина посчитаны сценарием S7, количества генов — простыми манипуляциями с хромосомной таблицей в Bash).
Более простые характеристики, приведенные в этом обзоре, получены как промежуточный этап при подготовке этих сценариев.
3. Результаты и обсуждение
3.1. Общая характеристика генома
Основные характеристики генома P. gaetbulicola приведены в табл. 1.
Для бактерий семейства Vibrionaceae характерны две различающиеся по размеру хромосомы; в литературе бо́льшую из них иногда обозначают «хромосома 1», а меньшую — «хромосома 2» (например, [4]). При этом в аннотации данного генома обозначения были противоположенными. В этом обзоре я придерживаюсь обозначения, использованного в аннотации генома — «хромосома 1» короче «хромосомы 2». Обратите на это внимание, чтобы избежать путаницы при сравнении с другой литературой.
| Длина, п.н. | Доля GC | Количество белок-кодирующих последовательностей | Доля белок-кодирующих последовательностей в молекуле | |
|---|---|---|---|---|
| Хромосома 1 | 2 052 529 | 47,9 % | 1 721 | 84,3 % |
| Хромосома 2 | 3 856 842 | 50,7 % | 3 321 | 85,1 % |
| Всего | 5 909 413 | 49,7 % | 5 042 | 84,8 % |
3.2. GC-skew. Ориджин репликации
Известно, что у большинства живых организмов количество мономеров гуанина примерно равно количеству мономеров цитозина на одной цепи; это же справедливо для аденина и тимина (второе правило Чаргаффа). Но это выполняется только для целых хромосом, в отдельных участках отношение количества различных нуклеотидов варьирует. Этот показатель характеризуют с помощью т.н. «skew» (англ. «перекос») (см. [10,11] и др.).
Для большей наглядности используют «cumulative» (англ. «накопительный») skew (см. «Материалы и методы»). У бактерий известно, что точка минимума cumulative GC-skew часто совпадает с ориджином репликации. Существует много гипотез, объясняющих это наблюдение (cм. введение в статье Tillier и Collins [11]).
Я построил диаграммы, иллюстрирующие cumulative GC- и AT-skew для хромосом рассматриваемой бактерии (рис. 1). Видно, что cumulative GC-skew формирует четкие минимумы и максимумы на каждой хромосоме. AT-skew образует не такой резкий, но хорошо заметный паттерн; его «знак» противоположен «знаку» GC-skew (это варьирует у разных бактерий [10,11]). Далее в тексте за ориджины репликации приняты точки минимума cumulative GC-skew (табл. 2). Координаты соответствующих пиков GC- и AT-skew отличаются на 0,1–1,7 % длины соответствующей хромосомы.
| Точка минимума cumulative GC-skew | Точка максимума cumulative AT-skew | Точка минимума cumulative GC-skew | Точка максимума cumulative AT-skew | |
|---|---|---|---|---|
| Хромосома 1 | 1 966 902 | 1 969 441 | 947 741 | 957 376 |
| Хромосома 2 | 325 535 | 308 499 | 2 219 789 | 2 153 172 |
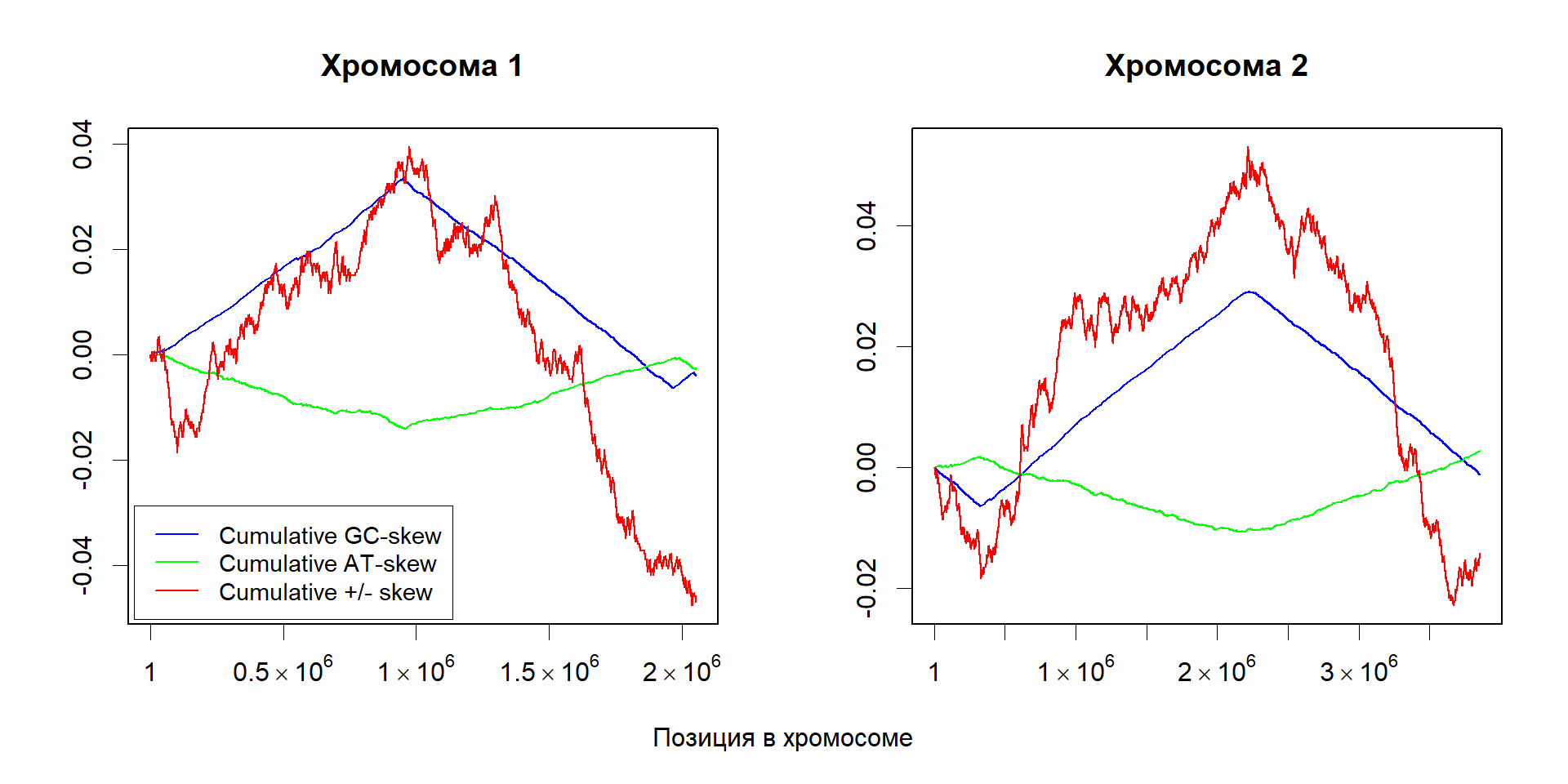
Известно, что гены у бактерий неравномерно распределены между лидирующей и отстающей цепями: больше генов смотрит от ориджина репликации (см. раздел 3.3.1). Можно представить себе ситуацию, в которой этого факта достаточно для объяснения GC- и AT-skew. Для того, чтобы закодировать протеом с данным аминокислотным составом, доступен определенный набор кодонов. Возможно, в этом наборе кодонов больше G, чем C, а в антикодонах, соответственно, наоборот. На одной половине цепи будет больше кодонов, а на другой — антикодонов, из-за чего и возникнет асимметрия в распределении G и C. Для того, чтобы проверить эту гипотезу, я построил диаграммы cumulative skew отдельно для кодирующих и некодирующих последовательностей (рис. 2).
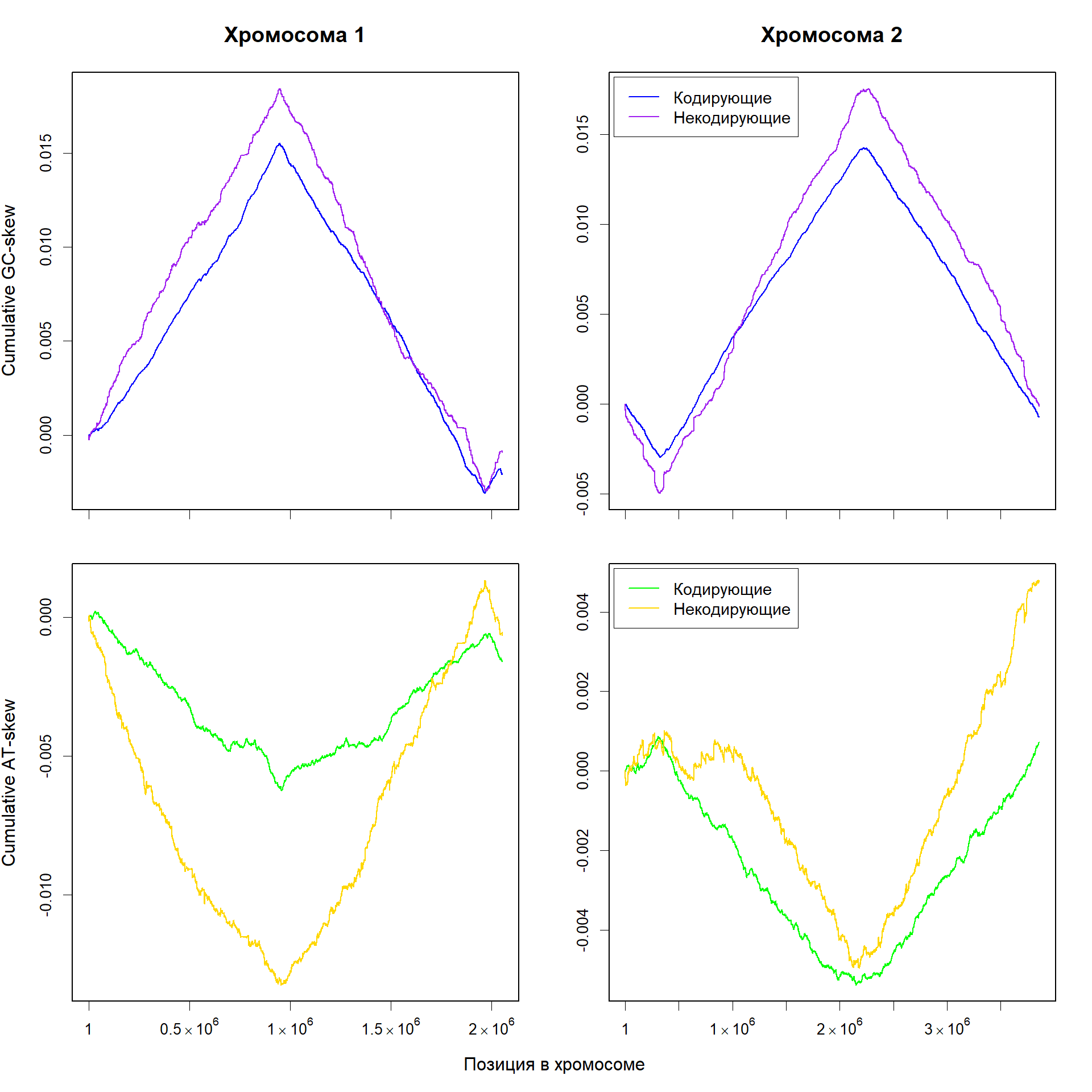
Очевидно, выдвинутая гипотеза не подходит как единственное объяснение: на некодирующих участках хромосомы skew выражен сильнее, чем на кодирующих. Это повторяет вывод Tillier и др. [11] на других бактериях.
3.3. Распределение генов белков
3.3.1. Направление генов относительно ориджина
Для того, чтобы визуализировать распределение генов белков между цепями хромосомы, я построили диаграмму, аналогичную той, которую обычно строят для cumulative GC-skew (рис. 1). К значению cumulative skew здесь прибавляется 1, если в этой позиции на хромосоме находится ген на «+» цепи, и вычитается 1, если на «−»; потом значения нормируются на количество генов на хромосоме.
Видно, что гены белков распределены асимметрично относительно ориджина репликации. Для хромосом 1 и 2 соответственно 55,4 и 57,5 % кодирующих белок последовательностей направлено от ориджина (т.е. закодировано на лидирующей цепи) (табл. 2). Это позволяет отвергнуть гипотезу о равновероятном нахождении гена на лидирующей или отстающей цепи с p-value = 7 × 10−6 и 4 × 10−18 соответственно.
Предположительно, это нужно для того, чтобы избежать столкновения «лоб в лоб» РНК-полимеразы и комплекса репликационной вилки [12]. Если это объяснение верно, можно предположить, что чем выше уровень экспрессии гена, тем выгоднее будет его направление от ориджина репликации, т.к. тем вероятнее РНК-полимераза будет находится на этом гене, когда до него дойдет репликационная вилка. Рибосомальные белки являются одними из самых высокоэкспрессируемых в бактериальной клетке, и, по крайней мере у E. coli, они действительно существенно чаще расположены так [12].
Сравним распределение по цепям отдельно рибосомальных, транспортных и «гипотетических» белков. Неохарактеризованные белки, скорее всего, будут довольно специфичными и низкокопийными, поэтому на них будет слабее действовать отбор, смещающий гены на отстающую цепь. Таким образом, можно ожидать уменьшение (приближение к 50 %) доли генов, направленных от ориджина, в ряду рибосомальные—транспортные—«гипотетические» белки.
| Гены белков | Гены рибосомальных белков | Гены транспортных белков | Гены «гипотетических» белков | |||||
|---|---|---|---|---|---|---|---|---|
| Всего | От ориджина | Всего | От ориджина | Всего | От ориджина | Всего | От ориджина | |
| Хромосома 1 | 1 721 | 954 (55 %) | 2 | 1 | 203 | 119 (59 %) | 275 | 148 (54 %) |
| Хромосома 2 | 3 321 | 1 911 (58 %) | 60 | 53 (88 %) | 302 | 154 (51 %) | 357 | 200 (56 %) |
Видно, что доля рибосомальных белков на лидирующей цепи действительно больше, чем среди остальных белков. Гипотеза с транспортными и «гипотетическими» белками не подтвердилась.
3.3.2. Расстояние от генов до ориджина
Очевидно, гены, находящиеся ближе к ориджину репликации, в среднем присутствуют в клетке в большем числе копий. Чем быстрее удваивается бактерия, тем сильнее выражен этот эффект.
Было показано, что в клетках бактерий (внутри одного вида) количество рибосом почти линейно возрастает с увеличением скорости роста [13]. Регуляция этого может осуществляться в т.ч. через расположение генов рибосомальных белков (и рибосомальных РНК — см. далее) на хромосоме.
Сравним среднее расстояние до ориджина репликации для рибосомальных, транспортных и «гипотетических» белков (табл. 3). Для всех групп генов (кроме двух генов рибосомальных белков на хромосоме 1) с помощью критерия χ2 оценим значимость отклонения распределения расстояний до ориджина от равномерного.
| Гены белков | Гены рибосомальных белков | Гены транспортных белков | Гены «гипотетических» белков | |
|---|---|---|---|---|
| Хромосома 1 | 0,258 | 0,120 | 0,248 | 0,257 |
| Хромосома 2 | 0,248 | 0,113 | 0,285 | 0,309 |
Если бы гены были равномерно распределены по хромосоме, можно было бы ожидать, что среднее расстояние до ориджина окажется равным ¼ длины хромосомы. Видно, что гены рибосомальных белков в среднем находятся значительно ближе к ориджину, что предполагает относительное увеличение их экспрессии при увеличении скорости роста и согласуется с литературой [9,13].
Также видно, что гены транспортных и «гипотетических» белков второй хромосомы находятся статистически значимо дальше от ориджина, чем можно было бы ожидать, если бы они были распределены равномерно. Это является некоторым свидетельством в пользу того, что их экспрессия уменьшается относительно экспрессии остальных генов при ускорении роста бактерии; спекулировать о биологической функции я не решаюсь.
3.4. Распределение генов транспортных и рибосомальных РНК
При увеличении скорости роста бактерии изменяется количественный состав не только белков, но и РНК в клетке. Например, оптимальное с точки зрения использования ресурсов клетки количество транспортных РНК на рибосому уменьшается [9]. В этой же работе показано, что отношение количества генов тРНК к количеству генов рРНК в геноме бактерии отрицательно коррелирует с максимальной характерной для нее скоростью роста: кроме того, гены как рРНК, так и тРНК в среднем расположены ближе к ориджину репликации у быстрорастущих бактерий, причем рРНК относительно ближе, чем тРНК.
Используя набор данных из статьи, я построил линейные регрессии в соответствующих осях и подставил в них данные для обсуждаемой бактерии (табл. 4).
| Характеристика | Значение | Оценка минимального времени удвоения по этой характеристике (в часах) |
|---|---|---|
| Количество генов тРНК | 136 | |
| Среднее расстояние от ориджина репликации до гена тРНК (в длинах хромосомы) | 0,179 | 1,55 |
| Количество генов рРНК | 29 | |
| Среднее расстояние от ориджина репликации до гена рРНК (в длинах хромосомы) | 0,098 | 1,83 |
| Разница между средним расстоянием от ориджина до гена тРНК и до гена рРНК | 0,081 | 1,29 |
| тРНК на рибосому в геноме | 14,07 | 1,92 |
Мы получаем довольно близкие значения минимального времени удвоения в 1,3–1,9 часа. Однако корреляция между характеристиками, по которым строилась регрессия, и временем удвоения хоть и значима, но не очень сильна [9], да и статистические методы я использовал топорно. Было бы интересно сравнить полученные так значения с настоящими, но мне не удалось найти в литературе информации о времени удвоения P. gaetbulicola. Было бы здорово, если бы получилось угадать таким способом хотя бы порядок.
4. Заключение
В работе представлен поверхностный обзор характеристик генома Photobacterium gaetbulicola Gung47. Некоторые из найденных особенностей (например, смещение от ориджина репликации генов транспортных и «гипотетических» белков на второй хромосоме), возможно, заслуживают более пристального рассмотрения.
Сопроводительные материалы
Материалы S1–S4, S7 — это сценарии на Python; S5, S6, S12 — сценарии на R.
Общее описание работы сценариев дано в разделе «Материалы и методы». Здесь приведены технические аспекты.
Сценарий S1 принимает на вход адрес хромосомной таблицы, вывод печатает в терминал.
Сценарий S2 принимает на вход адрес директории, в которой лежат файлы c названиями «chromosome_1.txt» и «chromosome_2.txt», содержащие последовательности хромосом 1 и 2 без дополнительных символов (приложены как файлы S8 и S9; сделаны из FASTA-файла с геномом с помощью Bash). Он создает в рабочей директории файл «cumulative_skew.txt» в котором по колонкам записаны координаты, значения cumulative GC-skew и значения cumulative AT-skew.
Сценарий S3 принимает на вход адрес хромосомной таблицы, затем адреса текстовых файлов с последовательностями хромосом 1 и 2 без дополнительных символов. В рабочей директории он создает папку «out» с четырьмя файлами, содержащими вывод программы: «chr1_cod.txt», «chr1_noncod.txt», «chr2_cod.txt», «chr2_noncod.txt». Они содержат координаты и значения GC-skew и AT-skew для соответственно кодирующих и некодирующих последовательностей первой и второй хромосомы.
Сценарий S4 принимает на вход путь до хромосомной таблицы и создает в рабочей директории файл с выводом.
Сценарий S5 принимает на вход пути к файлам с результатами работы сценариев S2 и S4 и создает изображение в рабочей директории.
Сценарий S6 принимает на вход путь до папки «out», являющейся результатом работы сценария S3, и в ней же создает изображение.
Сценарий S7 принимает на вход путь к хромосомной таблице, результат печатает в терминал.
Файл S8 — последовательность первой хромосомы бактерии без дополнительных символов.
Файл S9 — последовательность второй хромосомы бактерии без дополнительных символов.
Файл S10 — файл в формате FASTA, с полным геномом обсуждаемой бактерии, скачанный с FTP-сервера NCBI [6].
Файл S11 — хромосомная таблица обсуждаемой бактерии, скачанная с FTP-сервера NCBI [6].
Сценарий S12 принимает на вход адрес файла S13. Он выводит полученные значения времени удвоения (в часах) в следующем порядке: значение, основанное на среднем расстоянии от гена тРНК до ориджина репликации; на среднем расстоянии от гена рРНК до ориджина репликации; на разнице этих двух расстояний; на отношении количества генов тРНК и рРНК.
Файл S13 — это сопроводительный материал S5 из статьи Нu и др. [9], сохраненный в формате CSV.
Благодарности
Я благодарен факультету биоинженерии и биоинформатики и его преподавателям: Андрею Владимировичу Алексеевскому, Ивану Сергеевичу Русинову, Сергею Александровичу Спирину, Диме Босову и Дане Хлебникову за возможность написать эту работу.
Список литературы
- Kim, Y.O.; Kim, K.K.; Park, S.; Kang, S.J.; Lee, J.H.; Lee, S.J.; Oh, T.K.; Yoon, J.H. Photobacterium gaetbulicola sp. nov., a lipolytic bacterium isolated from a tidal flat sediment. Int. J. Syst. Evol. Microbiol. 2010, 60, 2587–2591, doi:10.1099/ijs.0.016923-0.
- Lau, N.-S.; Heng, W.L.; Miswan, N.; Azami, N.A.; Furusawa, G. Comparative Genomic analyses of the genus Photobacterium illuminate biosynthetic gene clusters associated with antagonism. Int. J. Mol. Sci. 2022, 23, 9712, doi:10.3390/ijms23179712.
- База данных NCBI Genome. Доступно на: https://www.ncbi.nlm.nih.gov/genome/?term=txid658445. Обращение 30.10.2022.
- Reen, F.J.; Almagro-Moreno, S.; Ussery, D.; Boyd, E.F. The genomic code: Inferring Vibrionaceae niche specialization. Nat. Rev. Microbiol. 2006, 4, 697–704, doi:10.1038/nrmicro1476.
- Vezzi, A.; Campanaro, S.; D’Angelo, M.; Simonato, F.; Vitulo, N.; Lauro, F.M.; Cestaro, A.; Malacrida, G.; Simionati, B.; Cannata, N.; и др. Life at depth: Photobacterium profundum genome sequence and expression analysis. Science 2005, 307, 1459–1461, doi:10.1126/science.1103341.
- NCBI FTP server. Доступно на: https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/940/995/GCF_000940995.1_ASM94099v1/. Обращение 9.10.2022.
- Новицкий, П.В.; Зограф, И.А. Оценка погрешностей результатов измерений; Миханова, В.Н., Ред.; 2-е изд.; Энергоатомиздат: Л., 1991; с. 191.
- Медицинская статистика. Доступно на: https://medstatistic.ru/methods/methods4.html. Обращение 24.12.2022.
- Hu, X.P.; Lercher, M.J. An optimal growth law for RNA composition and its partial implementation through ribosomal and tRNA gene locations in bacterial genomes. PLoS Genet. 2021, 17, 1–18, doi:10.1371/journal.pgen.1009939.
- McLean, M.J.; Wolfe, K.H.; Devine, K.M. Base composition skews, replication orientation, and gene orientation in 12 prokaryote genomes. J. Mol. Evol. 1998, 47, 691–696, doi:h10.1007/PL00006428.
- Tillier, E.R.M.; Collins, R.A. The contributions of replication orientation, gene direction, and signal sequences to base-composition asymmetries in bacterial genomes. J. Mol. Evol. 2000, 50, 249–257, doi:10.1007/s002399910029.
- Brewer, B.J. When polymerases collide: Replication and the transcriptional organization of the E. coli chromosome. Cell 1988, 53, 679–686, doi:10.1016/0092-8674(88)90086-4.
- Scott, M.; Gunderson, C.W.; Mateescu, E.M.; Zhang, Z.; Hwa, T. Interdependence of cell growth and gene expression: origins and consequences. Science 2010, 330, 1099–1102, doi:10.1126/science.1192588