Практикум 4
Комплексы ДНК-белок. Вторичная структура РНК
Задание 1
В этом задании предлагалось сравнить вторичную структуру тРНК, предсказанную программами einverted (emboss) и RNAfold (ViennaRNA, алгоритм Цукера) с реальной. PDB ID использованной тРНК — 1GTR.
Программа einverted находит локальные выравнивания данной последовательности с обратной комплементарной ей. Соответственно, ей нужны параметры, использующиеся при поиске выравнивания: "gap" (штраф за увеличение количества символов гэпа в выравнивании на один; отдельного штрафа за открытие гэпа нет), "match" и "mismatch". При этом почему-то "gap" записывается как положительное число, а "mismatch" — как отрицательное, хотя, очевидно, модуль того и другого берется с минусом при подсчете веса выравнивания. Кроме того, при запуске программы указывается порог — если нет ни одного выравнивания с весом выше порога, программа не возвращает ничего. Программа может вернуть несколько непротиворечащих друг другу выравниваний. При каждом запуске программы я указывал порог, равный нулю (самый низкий возможный). Если я говорю про "дефолтные параметры", я все равно подразумеваю, что порог равен нулю.
В задании предлагалось попробовать подобрать параметры, при которых вторичная структура предсказывается лучше всего. Я написал скрипт, который перебрал параметры: gap — от 0 до 12, match — от 0 до 10, mismatch — от 0 до −10. Так как порог равен нулю, запуски программы с параметрами, отличающимися пропорциально (например, 1:2:−2 и 2:4:−4), по сути эквивалентны, так как оптимальные выравнивания будут одинаковыми, и только их вес будет отличаться. Поэтому такие сочетания параметров, в которых наибольший общий делитель больше одного, я исключил.
Затем я сравнил каждое нашедшееся выравнивание с реальной структурой тРНК (pdb file → find_pair, см. пред. практикум). Для этого я вычислил балл: +1 за каждую правильно найденную каноническую пару, входящую в стебель, −1 за каждую неправильно найденную и −1 за каждую ненайденную (рис. 1).
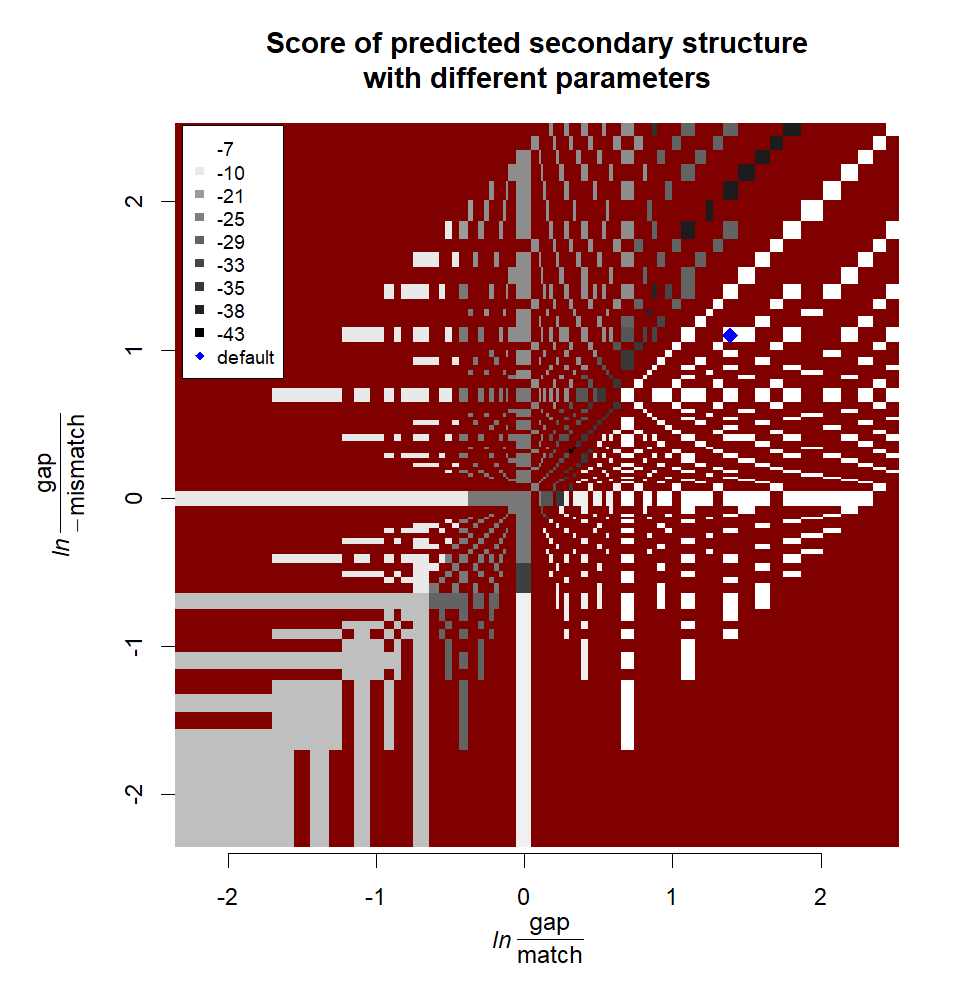
Во-первых, видно, что посчитанный так балл всегда получается отрицательным, т.е. ни одного выравнивания, действительно близкого к реальной структуре, не нашлось. Лучший результат — −7. Я проверил, все выравнивания с баллом −7 одинаковые.
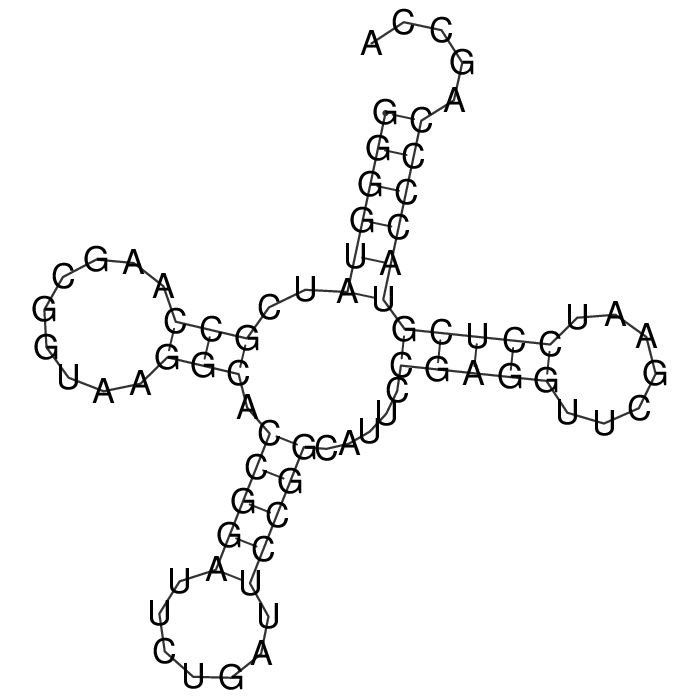
Чтобы можно было нагляднее сравнивать выравнивания, я записал их в dot-bracket notation и нарисовал с помощью программы RNAplot из пакета ViennaRNA (рис. 2, 3, 4). Лучшее предсказание (с баллом −7) — то, в котором нашелся только акцепторный стебель (рис. 3).
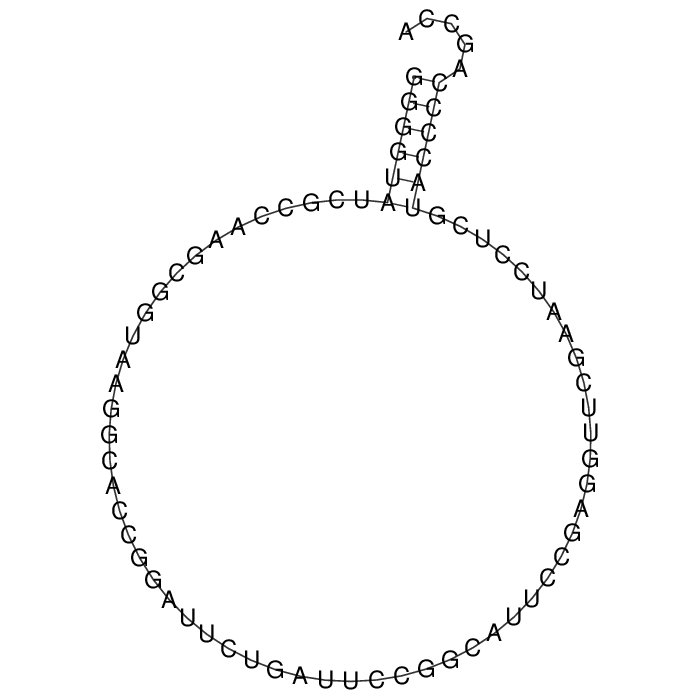
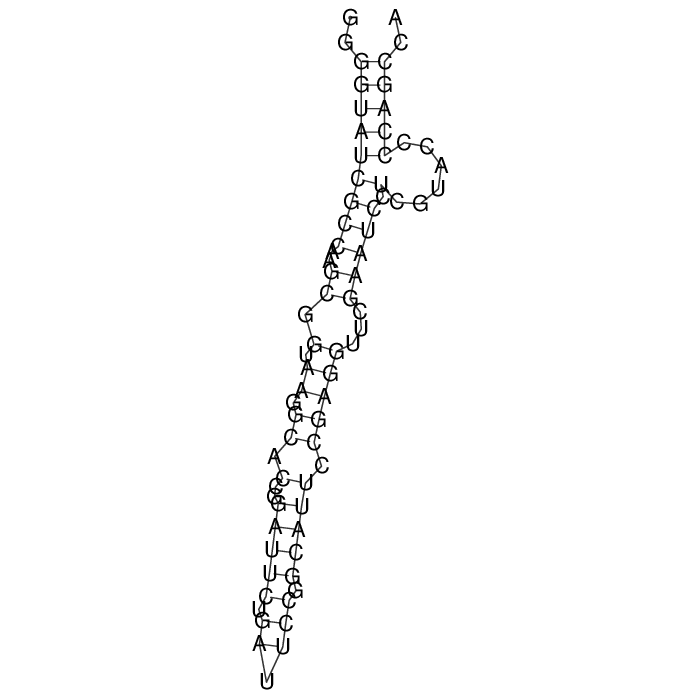
Программа RNAfold верно нашла все стебли, присутствующие в реальной структуре. Дополнительные водородные связи, не входящие в состав стеблей, она не нашла.
Я не думаю, что удастся найти параметры выравнивания, с которыми программа einverted предскажет вторичную структуру качественее, так как большую часть "разнообразия" возможных параметров я будто бы перебрал: остались непроверенными либо варианты, где какие-то из параметров отличаются многократно, больше чем в 10–12 раз ("за поля" на рис. 1), либо варианты с бóльшими значениями параметров, но похожие на те, которые уже есть (например, 10:20:−19 вместо 1:2:−2 — заполнение красного фона на рис. 1.) Почему-то мне кажется, что ничего из этого не приведет к принципальному улучшению качества предсказания.
| Участок структуры | Позиции в структуре (по результатам find_pair | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
|---|---|---|---|
| Акцепторный стебель | 2–7, 71–66 | 2–7, 71–66 | 2–7, 71–66 |
| D-стебель | 10–12, 25–23 | - | 10–12, 25–23 |
| Т-стебель | 49–53, 65–61 | - | 49–53, 65–61 |
| Антикодоновый стебель | 39–43, 31–27 | - | 39–43, 31–27 |
| Общее число канонических пар нуклеотидов | 20 | 6 | 19 |
Задание 2
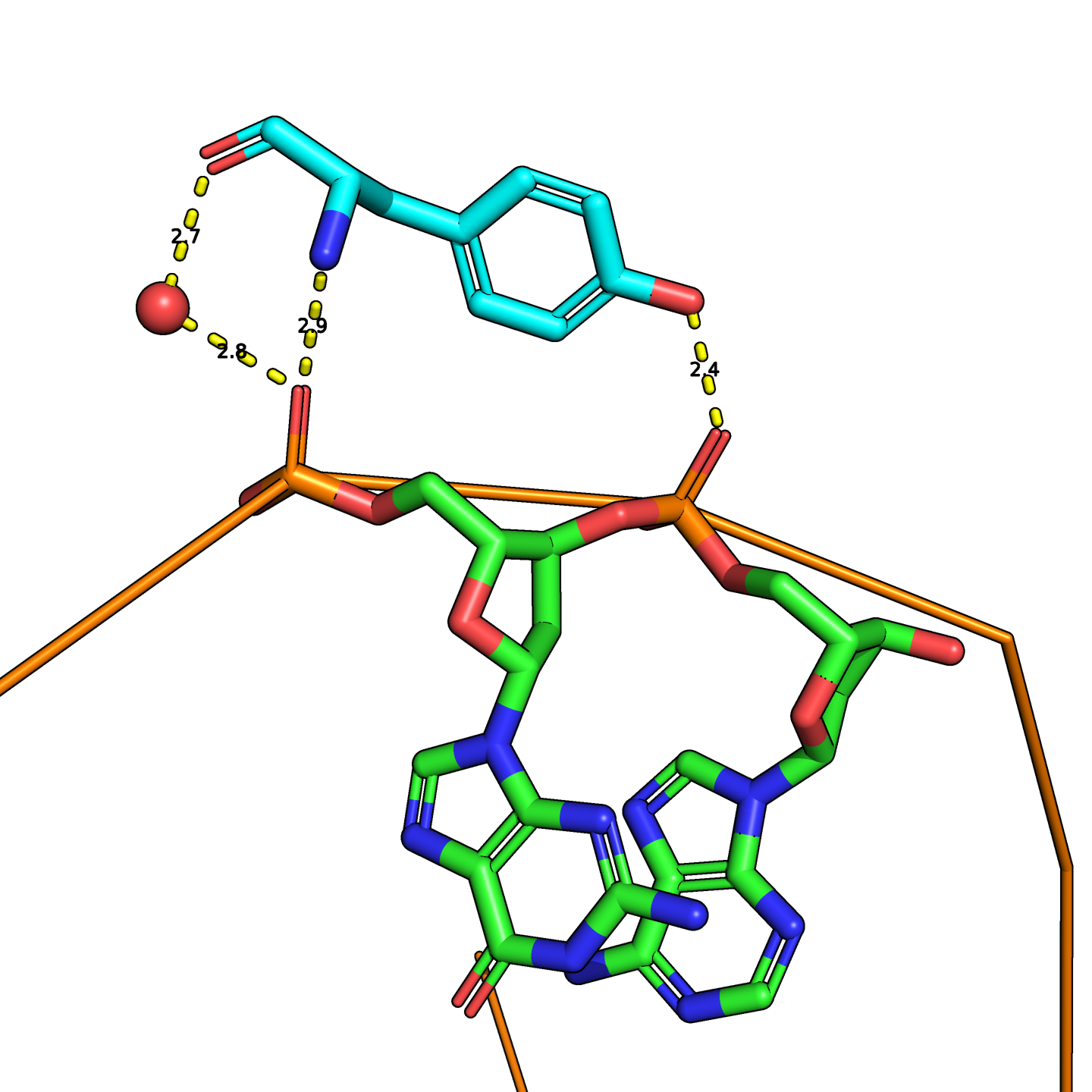
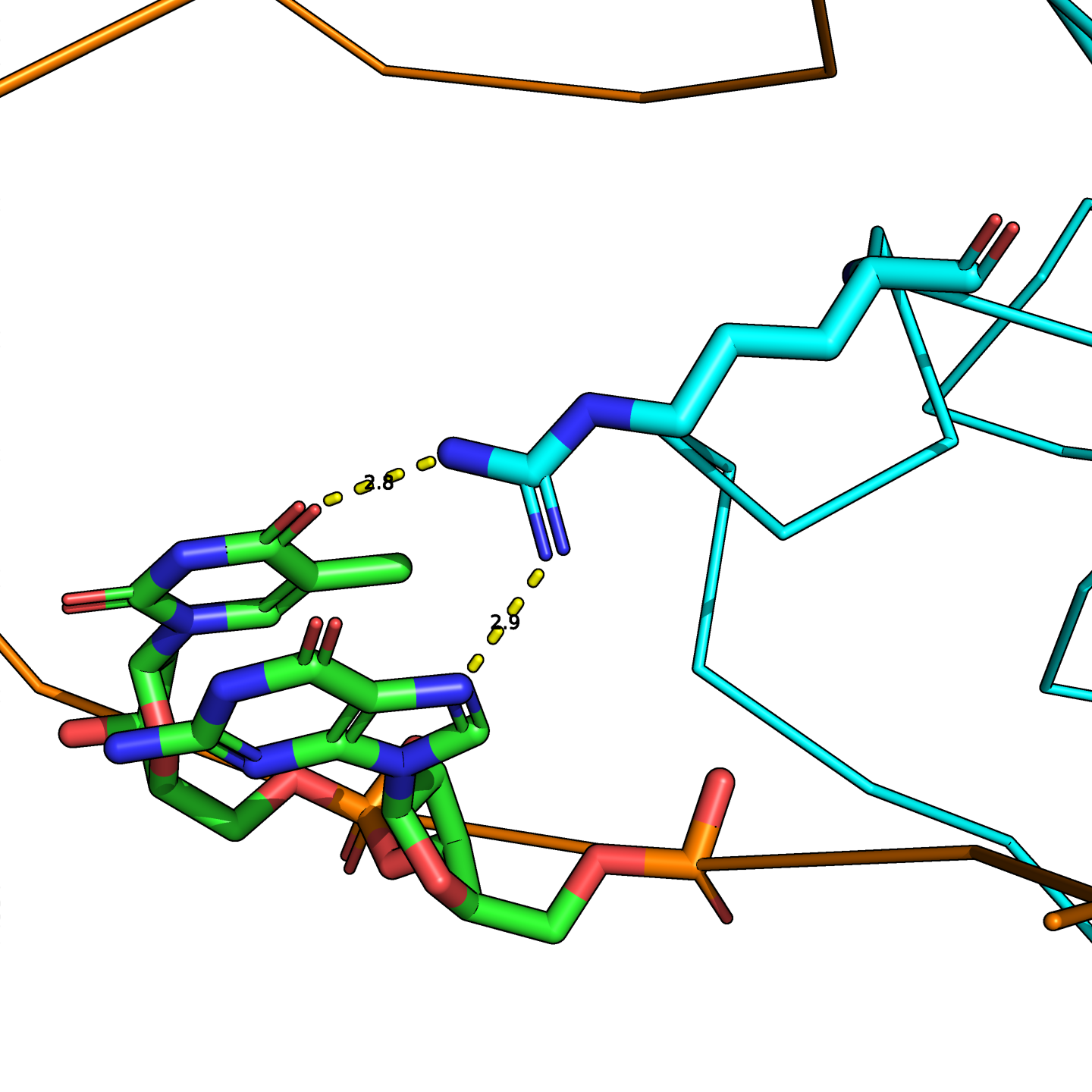
PDB ID структруы, рассматриваемой в этом задании — 1RH6.
Упражнение 1
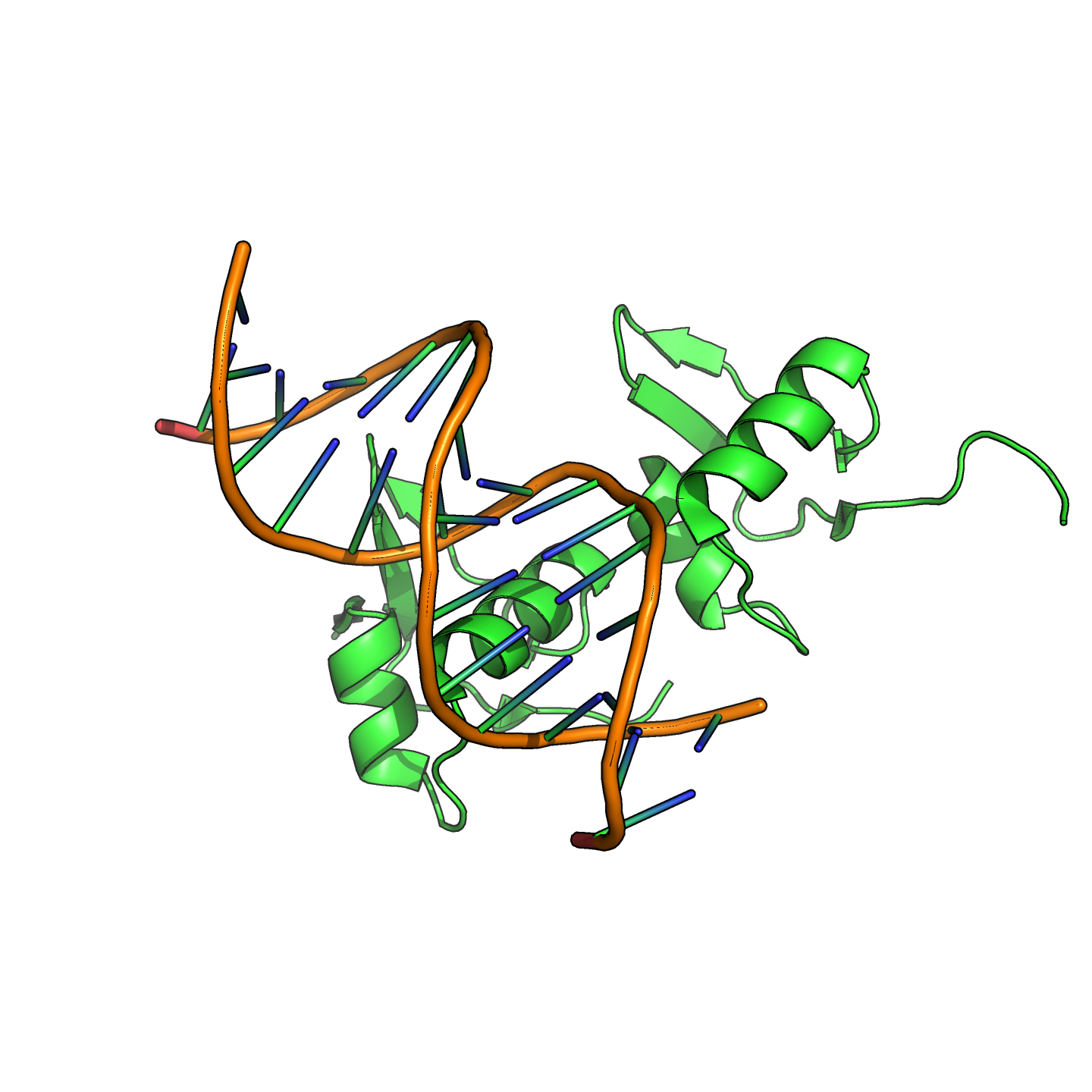
Упражнение 2
| Контакты атомов белка с: | Полярные контакты | Неполярные контакты | Всего |
|---|---|---|---|
| Остатками 2'-дезоксирибозы | 4 | 37 | 41 |
| Остатками фосфорной кислоты | 10 | 11 | 21 |
| Остатками азотистых оснований со стороны большой бороздки | 4 | 7 | 11 |
| Остатками азотистых оснований со стороны малой бороздки | 1 | 0 | 1 |
Как видно, неполярных контактов оказалось больше, чем полярных. Это ожидаемо: и неполярных атомов больше, и расстояние в определении "контакта" для них больше. Среди полярных выше доля контактов с остатками фосфорной кислоты, а среди неполярных — с остатками дезоксирибозы. Это тоже логично: в остатках фосфорной кислоты 2/3 атомов "полярные", а в остатках дезоксирибозы — 3/8. Кажется, никаких неожиданных результатов не получилось. И хорошо!
Упражнение 3
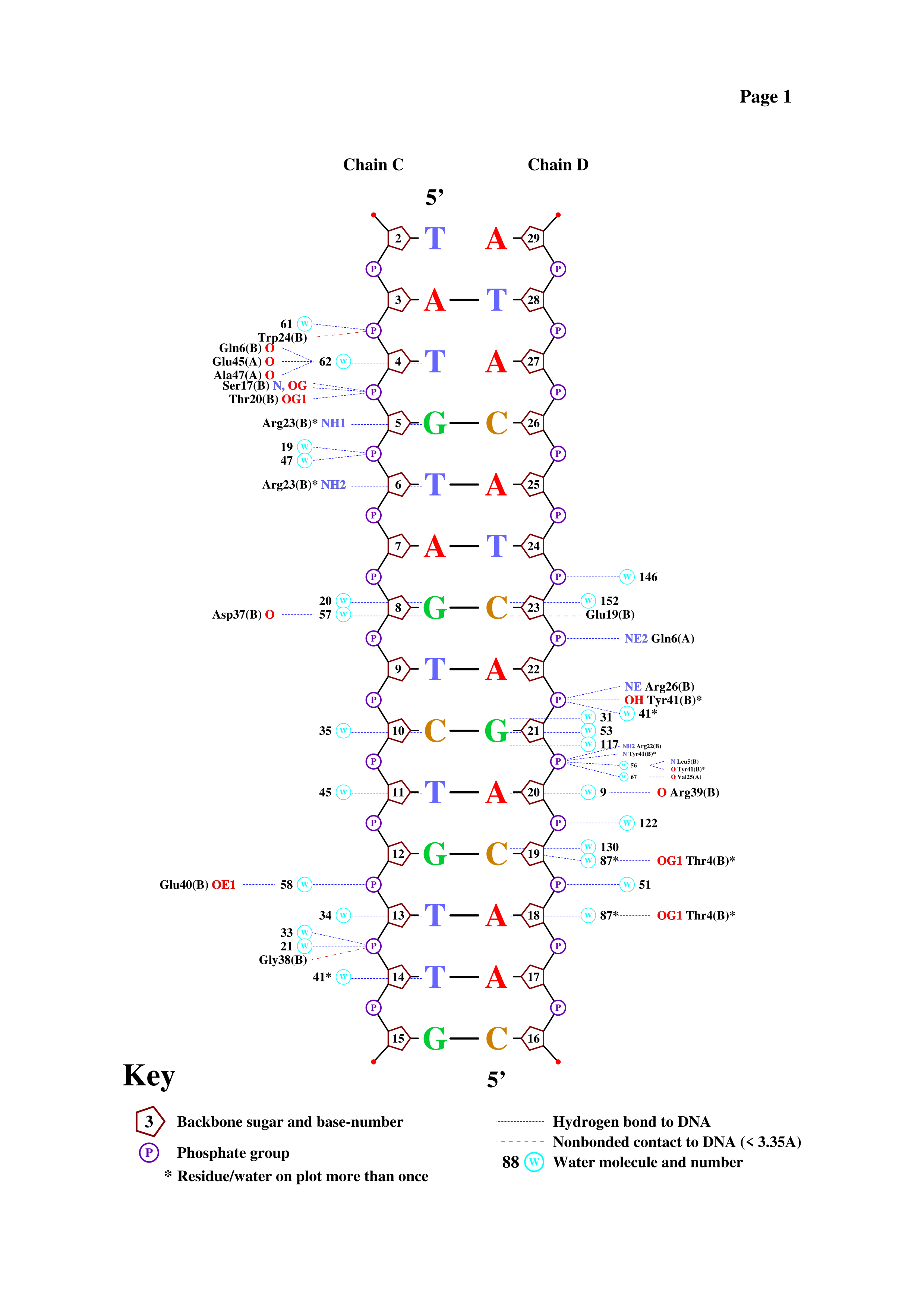
Упражнение 4