Практикум 8
Нуклеотидный BLAST
Задание 1
Чтобы выбрать подходящий фрагмент, я с помощью Emboss оставил скаффолды длиной 1000–10 000 п. н., а затем выбрал среди них один из тех, на которых аннотирована одна CDS. Идентификатор выбранного скаффолда в базе Nucleotide — NW_045521. Я не выделял из этого скаффолда более короткий фрагмент, а использовал его целиком (он 1280 п. н. длиной). На рис. 1 приведена схема аннотированных элементов на нем.
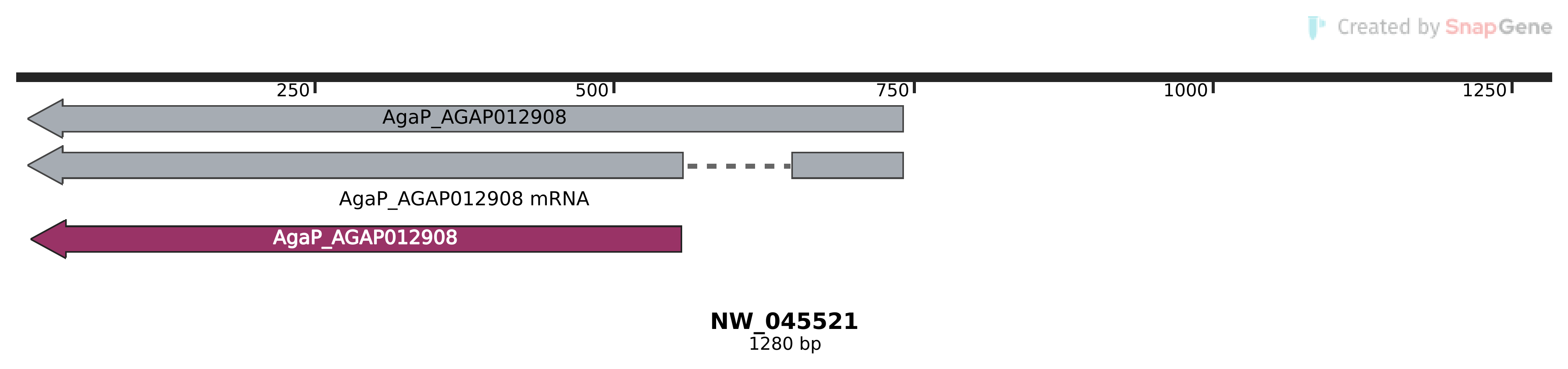
FASTA-файл с этим скаффолдом находится по ссылке.
Как таксон для сравнения я решил взять стрекоз — отряд Odonata, txid6961. С моим организмом — а это малярийный комар, Anopheles gambiae str. PEST, txid180454 — они расходятся на уровне инфракласса. И комары, и стрекозы относятся к подклассу крылатых насекомых (Insecta: Pterygota), но к разным инфраклассам: комары — новокрылые (Neoptera), а стрекозы — древнекрылые (Paleoptera). В задании в качестве примера приведены более таксономически далекие организмы, в частности, млекопитающих предлагается сравнить с птицами или амфибиями, т. е. с другими классами. Я решил взять стрекоз, потому что, если я верно это представляю, время дивергенции отрядов насекомых (кроме самых молодых, вроде бабочек и блох) примерно сравнимо со временем дивергенции классов тетрапод, так что, хотя они и ближе в ранговой классификации, сходство последовательностей ДНК должно быть сопоставимым.
Я пользовался BLAST на сайте NCBI. При запуске BLAST я всегда выставлял максимальное количество находок на 5000 — большое максимальное количество находок никак мешать не должно, а если оно будет меньше, чем реальное количество находок, то BLAST придется перзапускать. Кроме того, я выставил порог E-value, равный одному. На сайте NCBI по умолчанию он равен 0,05, что понятно — это принятый порог достоверности. Но на сайте EMBL-EBI он по умолчанию равен 10, и я подумал, что возьму "среднее" значение, котрое тоже кажется разумным: при значении меньше единицы мы считаем, что находка скорее неслучайна, чем случайна, хотя, возможно, и с не очень большой уверенностью. В общем, я могу себе представить, что какие-то находки с E-value между 0,05 и 1 могут представлять интерес. Кроме этих параметров я изменял длину слова для некоторых запусков.
Количество находок для разных запусков показано в табл. 1.
| Программа | Длина слова | Количество находок |
|---|---|---|
| megablast | 28 | 0 |
| 16 | 1 | |
| blastn | 11 | 4 |
| 7 | 6 | |
| blastx | 5 | 3 |
| 2 | 3 | |
| tblastx | 3 | 4 |
Три находки, нашедшиеся blastx с разной длиной слова, совпадают.
Теперь рассмотрим находки внимательнее.
Кажется, если бы я на самом деле искал последовательности, гомологичные данной, я бы использовал blastx, потому что она кодирующая, а использовать tblastx просто так (тем более в эукариотах) довольно странно. На рис. 2 показано graphic summary этих трех находок
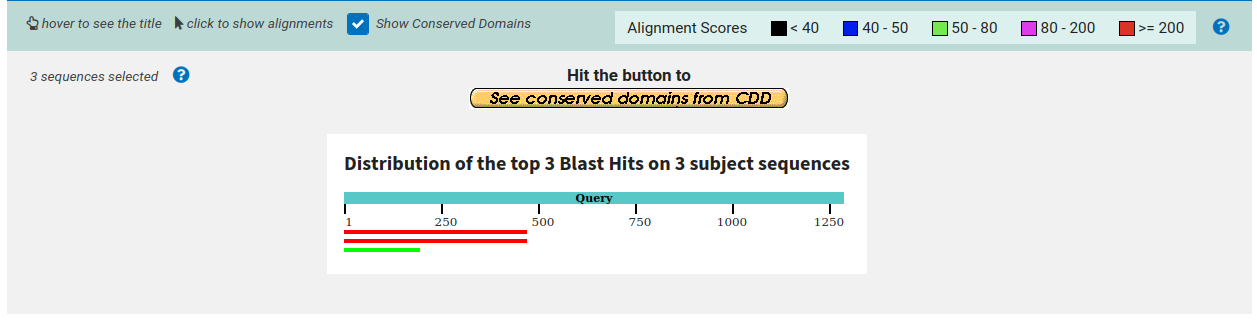
Все три находки очень значимы — E-value 6 × 10−70 — 2 × 10−15. Это находки в двух организмах: Ladona fulva (одна длинная находка) и Ischnura elegans (одна длинная находка и одна короткая). Две находки в Ischnura elegans — это две изоформы одного и того же белка, полученные с помощью автоматической аннотации. Для Ladona fulva нашедшийся белок указан как гипотетический, а для Ischnura elegans — как субъединица дегидролицилдифосфатсинтазного комплекса (катализирует этап синтеза какого-то изопреноида (Wikipedia)), причем внутри выравнявшегося фрагмента аннотирован известный каталитический домен. Находки расположены в начале нашей последовательности, то есть там, где находится кодирующая область. Короче, все выглядит так, будто мы действительно нашли гомологичные белки в двух стрекозах. Теперь сравним это с находками других алгоритмов.
tblastx сделал четыре находки, во всех из них несколько выравниваний (рис. 3). По сравнению с blastx, во-первых, он потерял гипотетический белок Ladona fulva, который в blastx был самой значимой находкой, E-value = 6 × 10−70 (!) Как такое могло произойти, мне не совсем понятно. Казалось бы, все, что нашел blastx, должен найти и tblastx, ведь в геноме есть ген данного белка. Да, он может быть прерван интронами, но выравниваем-то мы ген, а не другой белок, и он находится по белку, так что это не должно быть проблемой. Матрица выравнивания и штрафы у алгоритмов одинаковые, а длина слова у tblastx даже меньше. Непонятно.
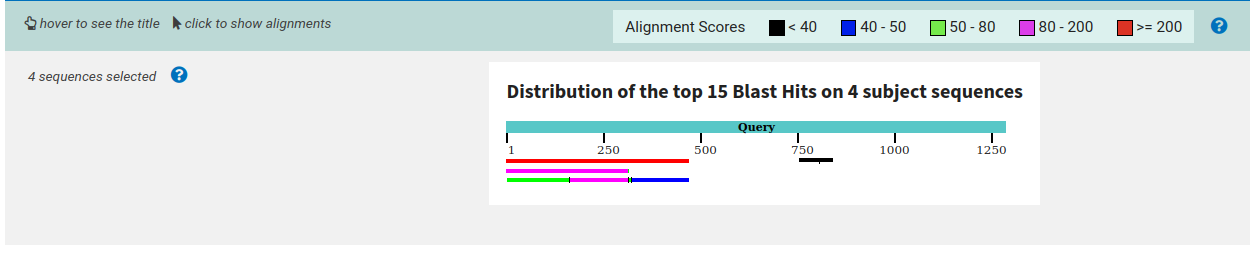
Дегидролицилдифосфатсинтазу Ischnura elegans он нашел, хотя теперь это выглядит менее аккуратно: много выравниваний про одно и то же.
Кроме того, tblastx нашел короткий участок в 20 нуклеотидов где-то в геноме Sympetrum striolatum с E-value = 0,78. Он попадает в область, которая в нашей последовательности аннотирована как некодирующая. Я не проверял, попадает ли туда какой-нибудь аннотированный элемент в геноме Sympetrum striolatum. Это находка кажется мне скорее случайной.
blastn нашел дегидролицилдифосфатсинтазу Ischnura elegans и, в зависимости от длины слова, один или три фрагмента в геноме Sympetrum striolatum длиной 27–44 нуклоетида с E-value 0,55 у всех. Среди них, кстати, нет фрагмента, найденного tblastx. megablast с длиной слова 16 нашел один из этих фрагментов, megablast с длиной слова 28 не нашел ничего.
В общем, мне кажется, что все находки в геноме Sympetrum striolatum случайны.
Для решения каких задач следует использовать какой алгоритм blast?
megablast следует использовать для поиска близких нуклеотидных последовательностей. Например, я хочу изучить популяционную структуру какого-нибудь вида и ищу все последовательности ITS, которые для него есть в базе данных. Сначала я поищу, конечно, просто с помощью Entrez, но потом, наверное, проверю megablast'ом, что ничего не пропустил.
blastn следует использовать для поиска более далеких нуклеотидных гомологов (как в следующем упражнении этого практикума).
blastx — для поиска гомологов кодирующих последовательностей (в типичном случае, как в этом упражнении), а tblastx — поиска гомологов кодирующих последовательностей в неаннотированных геномах.
Задание 2
В этом задании нужно было найти в геноме рассматриваемого эукариота гены рРНК по бактериальным последовательностям с помощью standalone blast.
Подготовка базы данных (на моем компьютере):
~/ncbi-blast-2.15.0+/bin/makeblastdb -in GCF_000005575.2_AgamP3_genomic.fna \ -dbtype nucl
Запуск blast:
~/ncbi-blast-2.15.0+/bin/blastn -task blastn -query rRNA_ecoli.txt \ -db GCF_000005575.2_AgamP3_genomic.fna -evalue 0.1 -word_size 7 \ -out blast_out_6.txt -outfmt 6
Использовал blastn c длиной слова 7 — это минимальная длина слова, которую можно выбрать в интерфейсе на сайте NCBI, и я подумал, что ее должно хватить для этой задачи. Выходной формат 6 — это TSV с находками. Можно скачать по ссылке.
Последовательности E. coli, которые я выравниваю — это последовательности 16S и 23S рибосомальных РНК. 16S входит в состав малой субъединицы и узнает последовательность Шайна — Дальгарно. 23S входит в состав большой субъединицы.
Для 16S нашлось 75 выравниваний, для 23S — 118. Дальше я посмотрел, были ли аннотированы участки, в которые попали находки, генами рРНК (я здесь и дальше пишу "гены", но имею в виду, конечно, не гены в строгом смысле, а кодирующие рРНК фрагменты генов, просто так короче):
grep '16S' blast_out_6.txt | cut -f 2,9,10 > 16S_hits grep '23S' blast_out_6.txt | cut -f 2,9,10 > 23S_hits ./tsv_to_usa.py 16S_hits 16S_hits.usa ./tsv_to_usa.py 23S_hits 23S_hits.usa extractfeat @16S_hits.usa -type rrna -describe gene -filter > rrna_16S_hits.txt extractfeat @23S_hits.usa -type rrna -describe gene -filter > rrna_23S_hits.txt infoseq rrna_16S_hits.txt -filter -columns N -delimiter ';' > 16S_hits_info.txt infoseq rrna_23S_hits.txt -filter -columns N -delimiter ';' > 23hits_info.txt cut -f 9 -d ';' 16S_hits_info.txt | tail -n +2 | sort -u cut -f 9 -d ';' 23S_hits_info.txt | tail -n +2 | sort -u
(Вот скриптик tsv_to_usa.py)
Вот выдача написанных выше команд для 16S:
[rRNA] (gene="5_8S_rRNA_a") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4NS0, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_b") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4PLY, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_d") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4QVV, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_f") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4S9Q, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_g") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4U21, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_h") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV52NW, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_i") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV52QR, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_j") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV52WU, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_l") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV53JW, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_m") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV551S, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_o") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_p") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_e") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4W1C, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_m") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_n") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV5735, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_p") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_q") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV5BWC, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_r") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence.
и для 23S:
[rRNA] (gene="5_8S_rRNA_b") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4PLY, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_c") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4PW5, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_e") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV4S13, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_i") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV52QR, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_k") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV53JJ, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_n") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV55JH, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_o") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="5_8S_rRNA_p") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_m") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_n") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV5735, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_p") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_q") Anopheles gambiae str. PEST chromosome Unknown CRA_x9P1GAV5BWC, whole genome shotgun sequence. [rRNA] (gene="SSU_rRNA_5_r") Anopheles gambiae str. PEST chromosome X, whole genome shotgun sequence.
Таким образом, для 16S нашлось 12 генов, аннотированных как 5,8S рРНК, для 23S — 8 генов, из них 4 общих. Генов, аннотированных как SSU (видимо, small subunit, т.е. 18S), нашлось соответсвенно 6 и 5, из них 5 общих. Все они расположены либо на хромосоме X, либо на неразмещенных скаффолдах.
Почему по 16S нашлись "SSU", понятно — они гомологичны. Почему по 23S нашлись 5,8S, тоже понятно — они гомологичны фрагментами. Но почему получилось и реципроктные находки, на первый взгляд понятно меньше. Кроме того, не понятно, почему не нашлись 28S. Чтобы лучше понять, что произошло, я посмотрел на рРНК, аннотированные во всем геноме, а не только в найденных фрагментах. Оказалось, что всего аннотировано 16 генов 5,8S рРНК, 19 генов "SSU рРНК" и два каких-то гена рРНК без более подробных обозначений в митохондриальном геноме(!) Ни одного гена 28S не аннотировано. Все аннотированные 5,8S имеют длину 158, что, кажется, нормально, а вот SSU имеют длину 496–878, что короче эукариотической 18S, и тем более 28S, так что что это такое, сразу сказать не получается. Было бы интересно порассматривать и те нашедшиеся фрагменты, в которых не было никакой аннотации, но на это, к сожалению, уже нет времени.