Рис 1. Расположение контига на геноме.
Скачаем архив с чтениями бактерии Buchnera aphidicola str. Tuc7 (идентификатор SRR4240378):
wget "ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/008/SRR4240378/SRR4240378.fastq.gz"
Для начала подготовим чтения с помощью программы trimmomatic, которую мы уже использовали в предыдущих практикумах.
Удалим возможные остатки адаптеров. Т.к. чтения непарные, используем TrimmomaticSE. Также создадим файл с последовательностями адаптеров: adapters.fasta (команда: cat /mnt/scratch/NGS/adapters/* > adapters.fasta).
TrimmomaticSE -phred33 SRR4240378.fastq.gz SRR4240378_trim.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7 2>logs.txt
В файле с логами есть строчка:
Input Reads: 4420587 Surviving: 4338744 (98.15%) Dropped: 81843 (1.85%)
Отсюда мы можем понять, что 1.85% чтений оказались адаптерами и были удалены.
Затем удалим правые концы с качеством ниже 20, и оставим только чтения не меньше 32 нуклеотидов длиной:
TrimmomaticSE -phred33 SRR4240378_trim.fastq.gz SRR4240378_trim_trim.fastq.gz TRAILING:20 MINLEN:32 2>logs_2.txt
В логах есть строка:
Input Reads: 4338744 Surviving: 4154738 (95.76%) Dropped: 184006 (4.24%)
Отсюда делаем вывод, что было удалено 4.24% чтений. До этой очистки было 4338744 ридов, а после стало 4154738.
Подготовим к-меры длиной 31 нуклеотид:
velveth . 31 -fastq.gz -short SRR4240378_trim_trim.fastq.gz
Параметр -short говорит о том, что чтения одиночные и короткие. На выход получаем несколько файлов, которые потребуются для дальнейшей сборки.
Запустим собственно саму сборку:
velvetg .
Как выход получаем несколько файлов, включая список контигов (contigs.fa), а также такую строку:
Final graph has 369 nodes and n50 of 7028, max 36746, total 657295, using 0/4154738 reads
Соответсвенно получается, что N50 = 7028
Посчитаем длины и покрытия трех самых длинных контигов:
cat contigs.fa |grep '^>' | sort -k4 -t '_' -nr | head -n 3
Получаем:
>NODE_8_length_36746_cov_20.017199
>NODE_57_length_19371_cov_20.546642
>NODE_15_length_16745_cov_20.901762
То есть самый длинный контиг имеет длину 36746 и покрытие 20.017199. Второй по длине - 19371 и покрытие 20.546642. Третий, длина - 16745, покрытие - 20.901762.
Найдем медиану покрытия:
cat contigs.fa |grep '^>' | wc -l
Получаем 208, то есть медиана - среднее значение покрытий 104ого и 105ого контигов:
cat contigs.fa |grep '^>' | sort -k6 -t '_' -nr | head -n 105 | tail -n 2
Получаем:
>NODE_79_length_6257_cov_18.495766
>NODE_115_length_1434_cov_18.413528
Таким образом, медианное покрытие - (18.495766 + 18.413528) / 2 = 18.454647
Посмотрим на контиги с аномально большим/маленьким покрытием (отличается от медианы не менее, чем в 5 раз):
Контиги с аномально высоким покрытием:
>NODE_81_length_934_cov_102.748390
>NODE_19_length_2106_cov_100.555084
Для меня удивительно, что контиги с самым большим покрытием, являются при этом достаточно длинными.
Контиги с аномально низким покрытием:
>NODE_166_length_64_cov_2.843750
>NODE_281_length_72_cov_2.944444
>NODE_271_length_31_cov_3.225806
>NODE_154_length_32_cov_3.593750
>NODE_294_length_63_cov_3.634921
При этом, контиги с маленьким покрытием - короткие.
Провдем BLASTN в NCBI, а точнее megablast, для каждого из трех самых длинных контигов и полным геномом Buchnera aphidicola (Sitobion miscanthi) strain LF. Выравниваем геном на контиг.
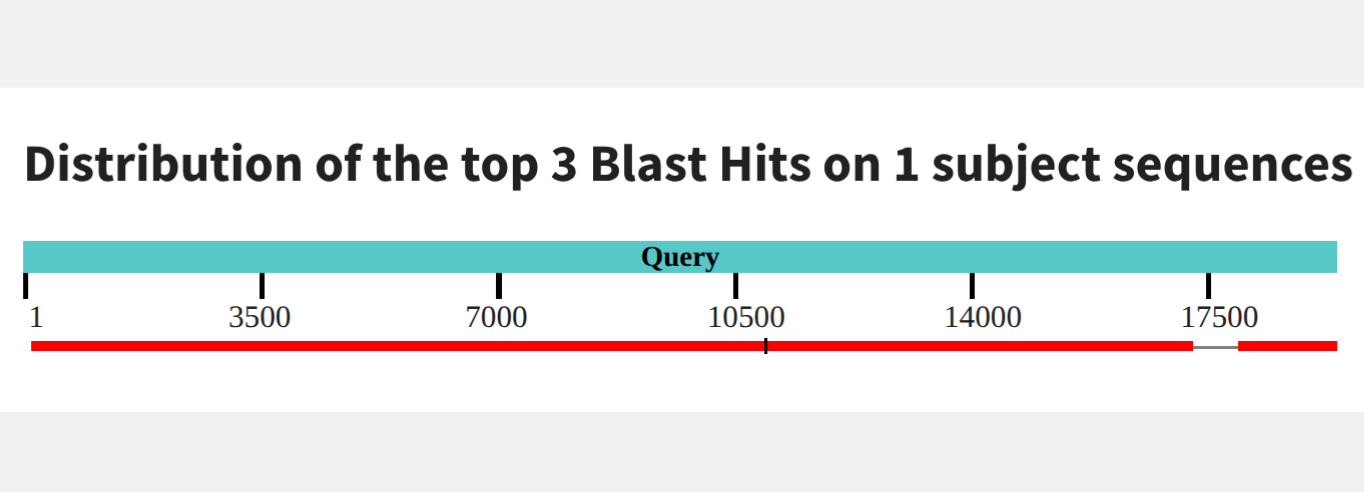
Самый длинный контиг (8):
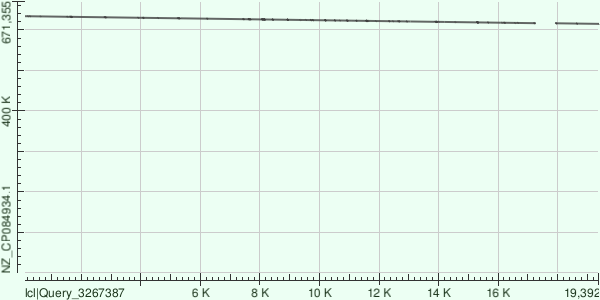
Получаем одно выравнивание из 2х частей (рис 1) с 84.39% процентов идентичности и карту локального сходства (рис 2).
Некоторые характеристики выравнивания:
Координаты участка хромосомы, соответствующего контигу - 521585-557768
Число однонуклеотидных различий - 5832
Число гэпов - 568
Какая доля контига выровнялась на геном - 84%
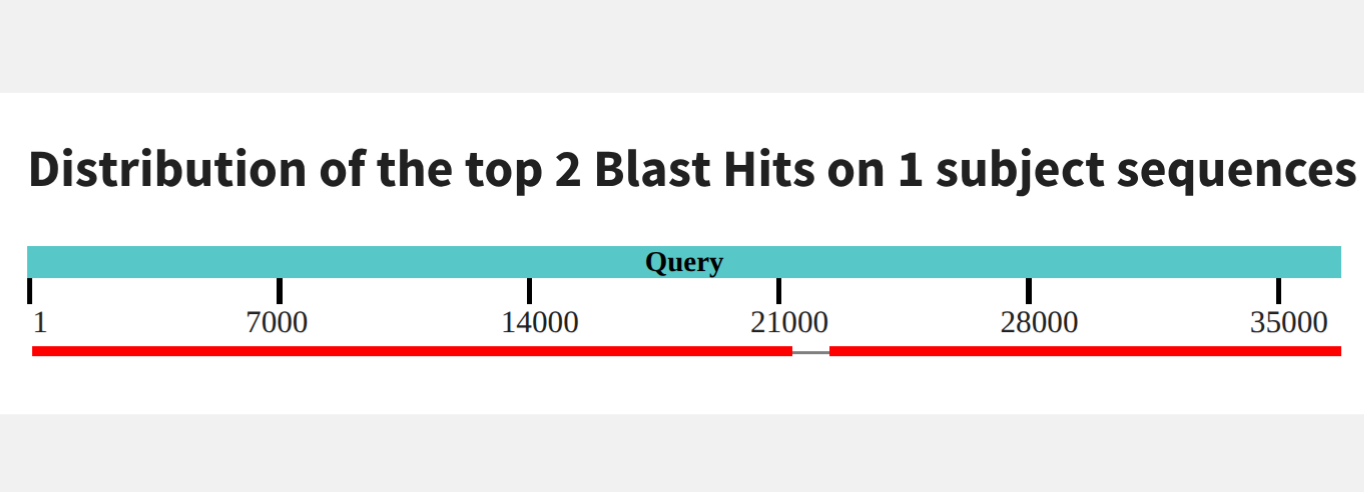
Второй по длине контиг (57):
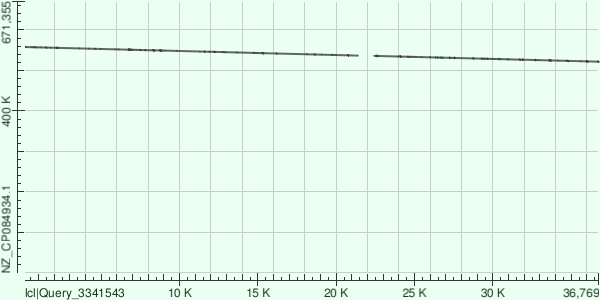
Получаем одно выравнивание (рис 3) с 84.45% процентов идентичности и карту локального сходства (рис 4).
Некоторые характеристики выравнивания:
Координаты участка хромосомы, соответствующего контигу - 142890-159525
Число однонуклеотидных различий - 2601
Число гэпов - 181
Какая доля контига выровнялась на геном - 84%
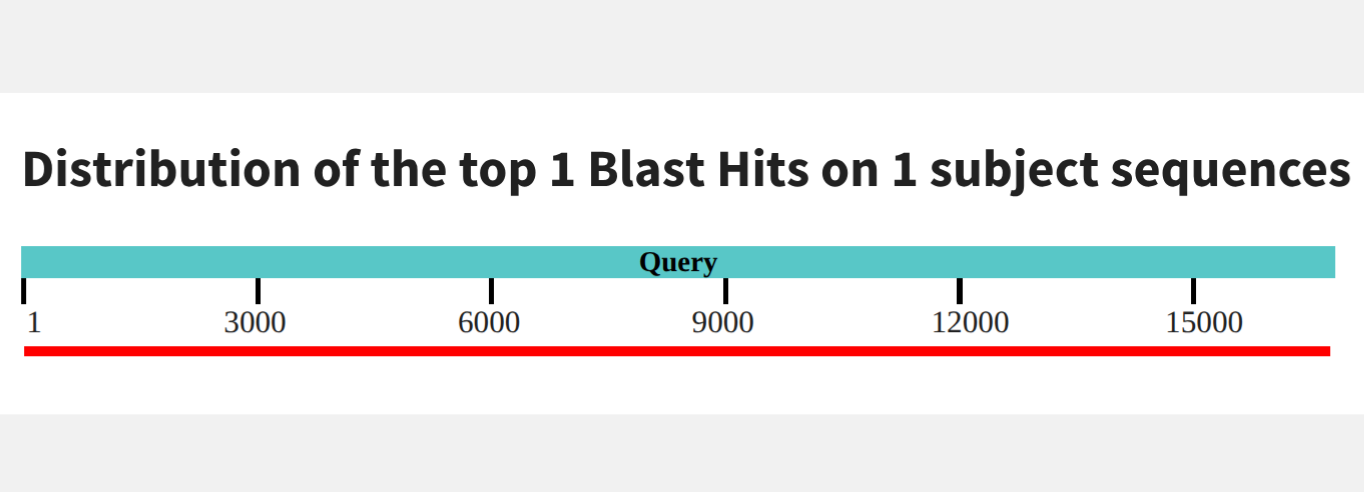
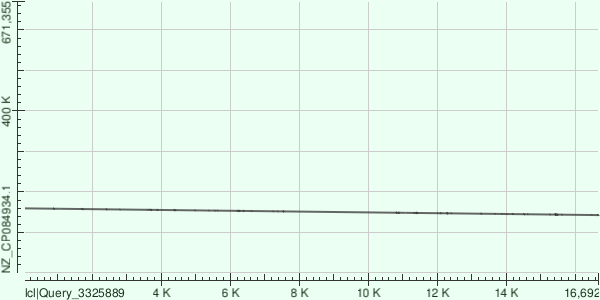
Третий по длине контиг (15):
Получаем одно выравнивание из 3х частей (рис 5) с 83.94% процентов идентичности и карту локального сходства (рис 6).
Некоторые характеристики выравнивания:
Координаты участка хромосомы, соответствующего контигу - 614905-633838
Число однонуклеотидных различий - 3054
Число гэпов - 274
Какая доля контига выровнялась на геном - 84%