Создаем список из идентификаторов цитохромов В из выбранных животных.
С помощью EMBOSS создаем fasta файл с последовательностями этих белков. Команда:
seqret @cyb.list cyb.fasta
Выравниваем белки программой muscle. Команда:
muscle -align cyb.fasta -output cyb-alignment.fasta
С помощью библиотеки BioPython переведем выравнивание в формат, который может воспринять программа FastME, а именно в формат phylip-relaxed:
from Bio import AlignIO
inh = open("cyb-alignment.fasta", "r")
outh = open("cyb.phy", "w")
alignment = AlignIO.parse(inh, "fasta")
AlignIO.write(alignment, outh, "phylip-relaxed")
inh.close()
outh.close()
Теперь проведем реконструкцию, выберем в качестве модели p-distance:
fastme -i cyb.phy -o p-distance_cyb.phy -pp
Снова проведем реконструкцию, но уже в качестве модели возьмем MtREV:
fastme -i cyb.phy -o MtREV_cyb.phy -pM
Проведем еще одну реконструкцию, с помощью программы iq-tree:
iqtree -s cyb.phy
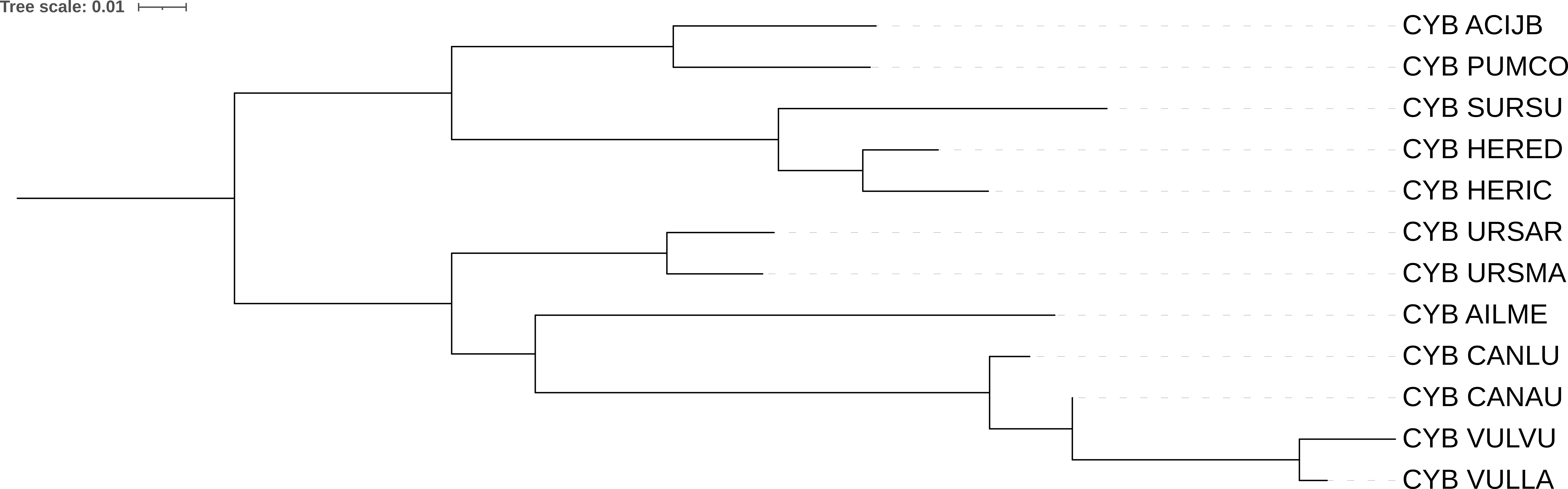
Будем сравнивать наши деревья с таксономическим деревом (рис 1).
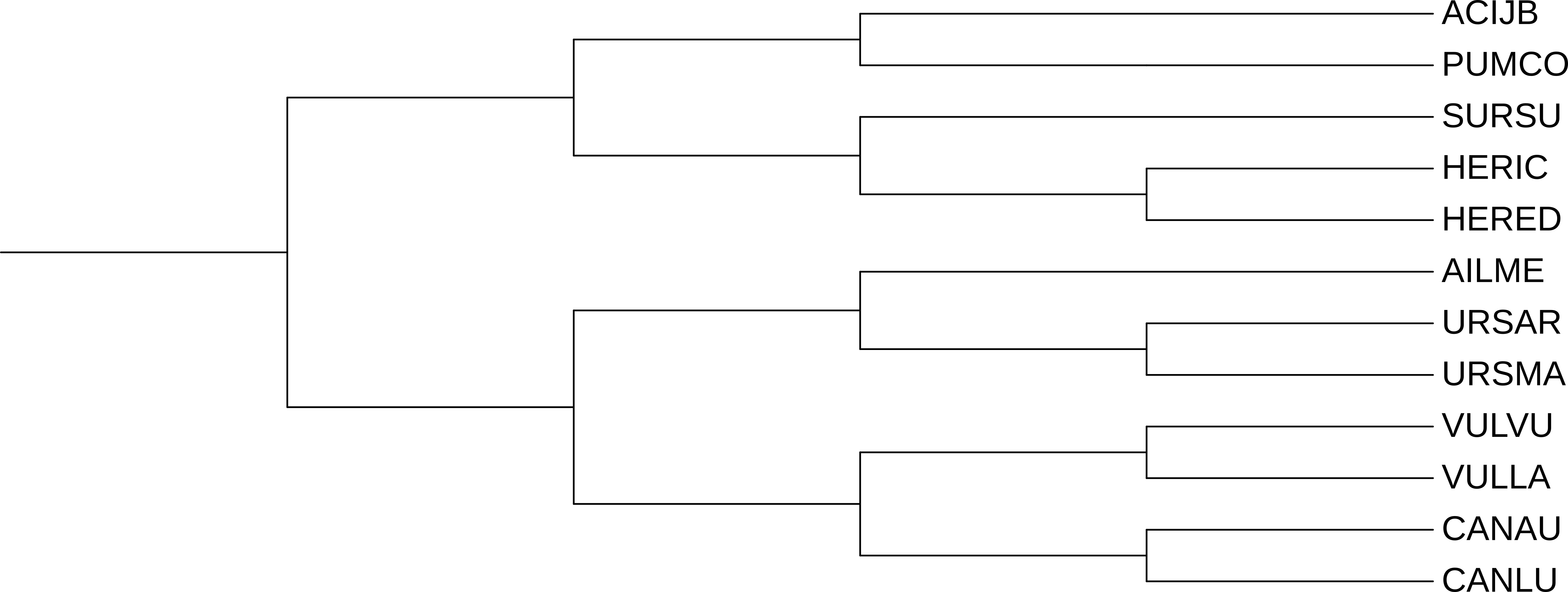
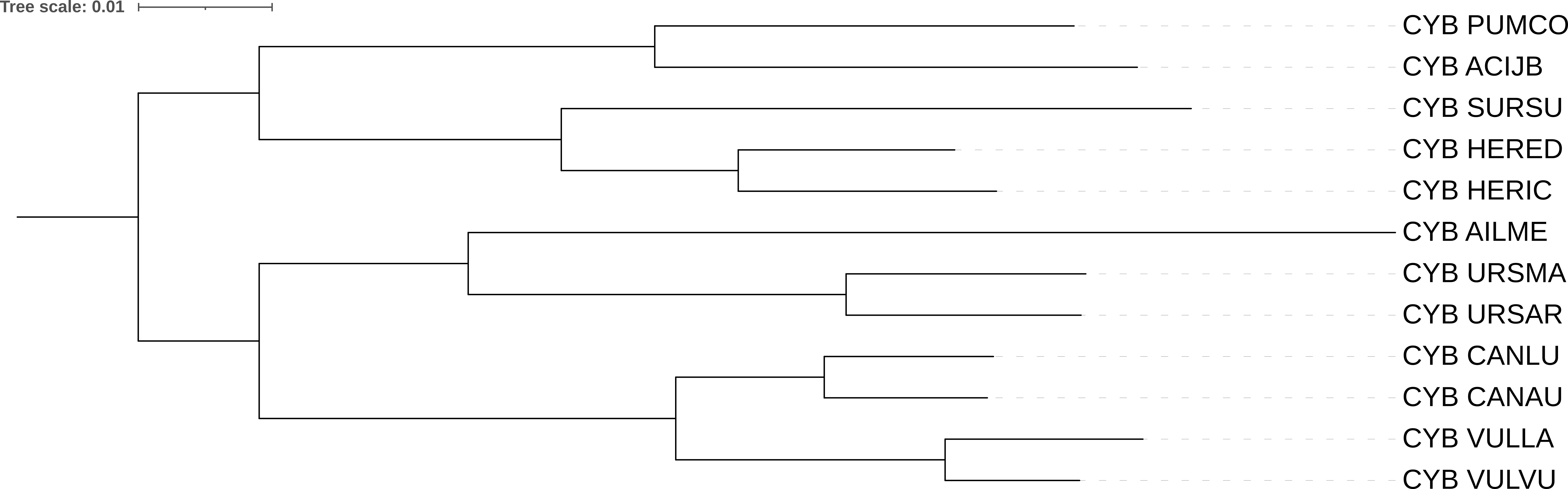
Моделью p-distance (рис 2) и моделью MtREV (рис 3) дерево реконтруировалось идеально, топологически все ровно так же, как и в таксономическом дереве.
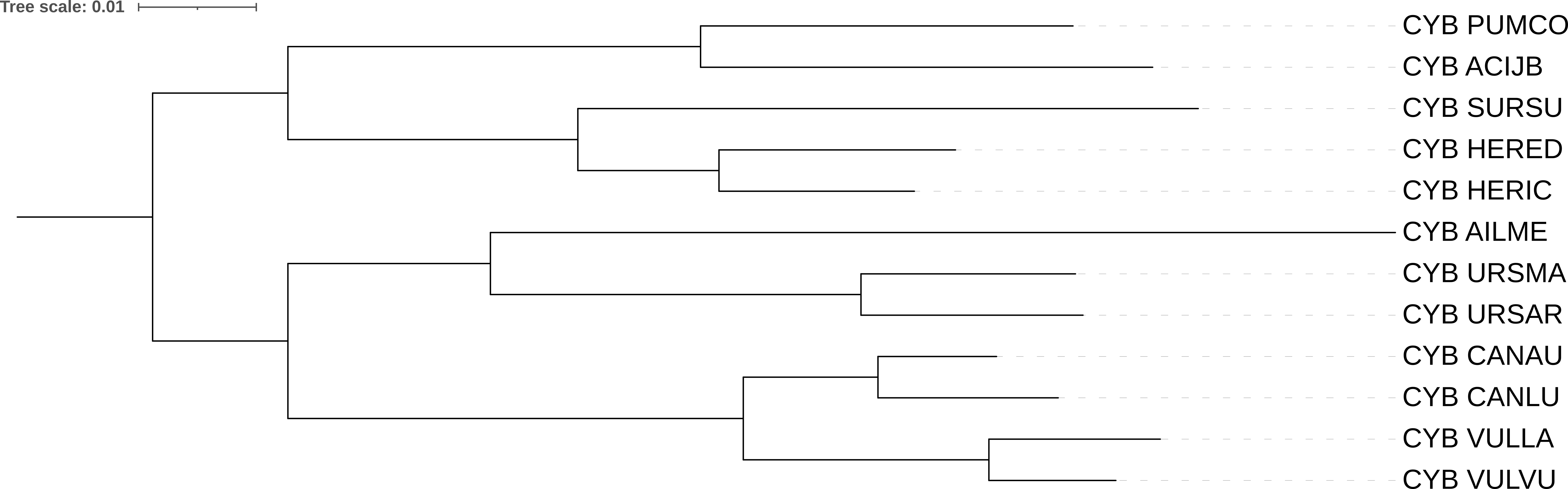
Моделью iqtree (рис 4) дерево также реконструировалось хорошо, но отсутвует клада (CANLU, CANAU). Вместо этого CANAU является сестринской к кладе (VULVU, VULLA), а CANLU уже является сестринской к кладе из всех трех, обсуждаемых ранее организмов. Также, хочется отметить, что ветвь ведущая к листу CANAU получилась длины 0.
С положением AILME (большая панда) тоже есть вопросы. В таксономическом дереве, этот организм должен быть сестринским к кладе (URSAR, URSMA) (это медведи), а CANLU, CANAU, VULVU и VULLA (то есть волки и лисы) образуют отдельную ветку. Здесь же, AILME находится в другой ветке нежели медведи. Он объединяется в кладу с волками и лисами.