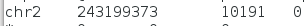|
Команда
|
Значение
|
|
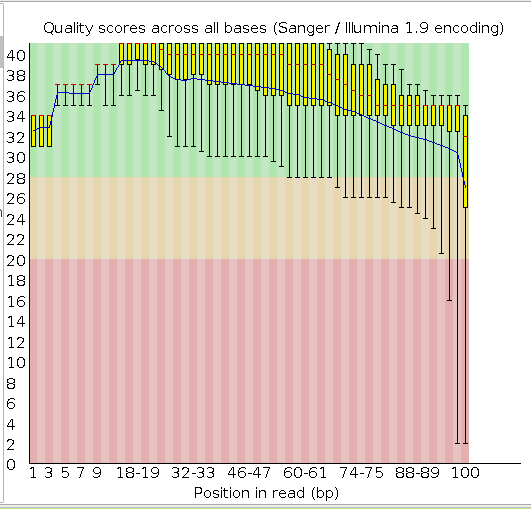
fastqc chr2.fastq
|
Анализ качества чтений
|
|
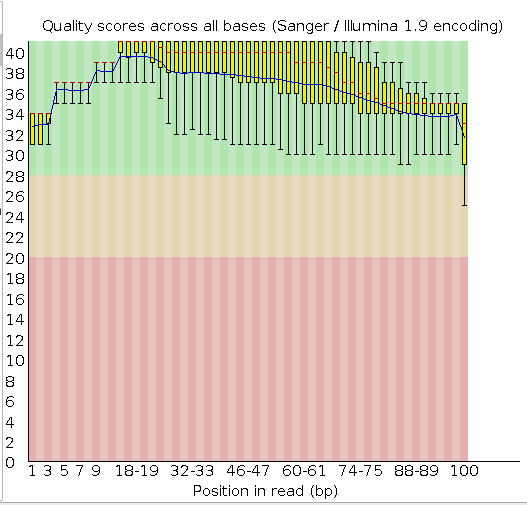
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr2.fastq chr2t.fastq TRAILING:20 MINLEN:50
|
Удаляет чтения с плохим качеством по phred33 или обрезает их.
|
|
bwa index chr2.fasta
|
Индексирует референсную последовательность.
|
|
bwa mem chr2.fasta chr2t.fastq > chr2_exom.sam
|
Картирует чтения на референсную последовательность, получается файл в формате sam.
|
|
samtools view -bo chr2_exom.bam chr2_exom.sam
|
Переводит данные о картированных чтениях из формата sam в формат bam(бинарный).
|
|
samtools sort chr2_exom.bam chr2_exom.sorted
|
Сортирует картированные чтения по координате начала чтения в референсе.
|
|
samtools index chr2_exom.sorted.bam
|
Индексирует сортированный файл.
|
|
samtools idxstats chr2_exom.sorted.bam
|
Показывает, сколько чтений откартировалось на референс.
|
|
samtools mpileup -uf chr2.fasta chr2_exom.sorted.bam > chr2_exom_SNPs.bcf
|
Создаёт файл с информацией об SNP
|
|
bcftools call -cv chr2_exom_SNPs.bcf -o chr2_exom_SNPs.vcf
|
Конвертирует файл в формате bcf в файл в формате vcf со списком отличий между референсом и чтениями.
|
|
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr2_exom_SNPs.vcf > chr2_exom_SNPs.avinput
|
Переводит файл с заменами из формата vcf в формат avinput, с которым может работать программа annovar.
|
perl /nfs/srv/databases/annovar/annotate_variation.pl --geneanno -out refGene -build hg19 -dbtype refGene chr2_exom_SNPs.avinput /nfs/srv/databases/annovar/humandb/
|
Аннотирует файл с SNP по базе данных генов refGene.
|
|
perl /nfs/srv/databases/annovar/annotate_variation.pl --filter -out dbsnp -dbtype snp138 -build hg19 chr2_exom_SNPs.avinput /nfs/srv/databases/annovar/humandb/
|
Аннотирует файл с SNP по базе dbsnp.
|
|
perl /nfs/srv/databases/annovar/annotate_variation.pl --filter -out 1000genomes -dbtype 1000g2014oct_all -build hg19 chr2_exom_SNPs.avinput /nfs/srv/databases/annovar/humandb/
|
Аннотирует файл с SNP по базе 1000genomes.
|
|
perl /nfs/srv/databases/annovar/annotate_variation.pl --filter -out gwas -dbtype gwasCatalog -build hg19 chr2_exom_SNPs.avinput /nfs/srv/databases/annovar/humandb/
|
Аннотирует файл с SNP по базе GWAS.
|
|
perl /nfs/srv/databases/annovar/annotate_variation.pl --filter -out clinvar -dbtype clinvar_20150629 -build hg19 chr2_exom_SNPs.avinput /nfs/srv/databases/annovar/humandb/
|
Аннотирует файл с SNP по базе Clinvar.
|