Сборка генома de novo
Введение
В этом практикуме мы будем работать с сиквенсом генома бактерии Buchnera Buchnera aphidicola, штаммом str. Tuc7, который живёт в симбиотических отношениях с видом Гороховой тли (Acyrthosiphon pisum). Buchnera aphidicola, грам-отрицательная бактерия, представитель Pseudomonadota и единственный вид рода Buchnera. Взаимоотношения между тлей и бактерией - мутуализм, и ни один из взаимодействующих организмов не может размножаться независимо. Buchnera - близкий родственник E.coli, но Buchnera содержит более 100 копий генома в клетке. Имеет достаточно короткий геном от 600 до 650 kb, кодирующий примерно 500-560 белков.
В этом практикуме мы хотим попробовать собрать геном данного штамма из коротких чтений длины 39, полученных по технологии Illumina — SRR4240359
Обрезка некачественных фрагментов у прочтений
Удалим адаптеры. Для этого объединим их все адаптеры в один файлcat /mnt/scratch/NGS/adapters/*.fa > all_adapters.fastaПосчитаем количество адаптеров:
grep -vc '^>' all_adapters.fastaУдалим адаптеры из чтений:
TrimmomaticSE -phred33 SRR4240359.fastq.gz trimmed.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7Выдача идёт в файл trimmed.fastq.gz и в командную строку:
Input Reads: 13'557'938 Surviving: 13'502'066 (99.59%) Dropped: 55'872 (0.41%)
То есть на вход триммонатик принял 13'557'938 и отсеял от них 55872 (0.41%). Это и есть адаптеры illumina
Отрежем нуклеотиды с конца чтений, если их качество меньше 20 и отсеим чтения короче 32:TrimmomaticSE -phred33 trimmed.fastq.gz cutted.fastq.gz TRAILING:20 MINLEN:32Аналогиично выдача в файл cutted.fastq.gz и в командную строку:
Input Reads: 13'502'066 Surviving: 12'184'080 (90.24%) Dropped: 1'317'986 (9.76%)
Тут триммоматик отсеял 1'317'986 (9.76%) от 13'502'066 чтений.
Можем посмотреть размер файлов с помощью командыdu -h <путь к файлу>
445M весит изначальный файл с прочтениями 443M весит файл с выравниваниями после удаления адаптеров 385M весит файл с обрезанными выравниваниями
Сборка генома с помощью velvet
Сначала воспользуемся velveth, чтобы разбить наши прочтения на k-меры, из которых потом, с помощью графа де-Брёйна, velvetg будет собирать геном.
velveth output_velvet 31 -short -fastq.gz cutted.fastq.gzoutput_velvet — папка, куда velveth положит три файла:
- Log информация о запуске и об ошибках
- Roadmaps бинарный файл, содержащий информацию о маршрутах ридов через граф де-Брёна
- Sequences получившиеся k-меры
velvetg output_velvetВ output_velvet появились:
- contigs.fa файл fasta содержит последовательности контигов длиной более 2k, где k - длина слова, используемая в velveth. Их (grep -c "^>" contigs.fa) 285
- PreGraph, Graph, LastGraph по сути три технических файла трёх разных состояний графа де-Брёйна.
- stats.txt таблица с информацией о контигах и др. Чтобы получить длину в нуклеотидах каждого узла, вам просто нужно добавить k − 1, где k - длина слова, используемая в velveth.
Также в stats.txt есть чтолбцы *cov и *Ocov. Разница между ними заключается в способе вычисления этих значений. При первом подсчете к сумме покрытия добавляются слегка отличающиеся последовательности. Однако при втором, более строгом подсчете, учитываются только те последовательности, которые идеально соответствуют согласованной последовательности.
Анализ получившихся контигов
N50 = 125674 c покрытием 44,6%. Вот три самых длинных контига:
grep '^>' contigs.fa | awk -F'_' '{print $1, $2, $3, $4, $5, $6}' | sort -k4 -nr | head -n 3 | less
>NODE 11 length 125674 cov 44.550949 >NODE 1 length 108447 cov 42.009186 >NODE 14 length 71403 cov 39.411552
Поиск медианы и аномальных покрытий
Всего найдено 285 контигов (grep -c "^>" contigs.fa), медианным будет 143
grep '^>' contigs.fa | awk -F'_' '{print $1, $2, $3, $4, $5, $6}' | sort -k6 -nr | tail -n143 | head -n1
>NODE 342 length 68 cov 5.970588
Медиана получилось небольшая. Из-за этого статистически будут только аномально большие покрытия:
grep '^>' contigs.fa | awk -F'_' '{print $1, $2, $3, $4, $5, $6}' | awk '$6 > 29.85' | wc -l
52
>29.85 - мы отбираем контиги, длина которых превышает покрытие медианы более чем в 5 раз. Самое большое покрытие - >NODE 98 length 47 cov 139.489365 - покрытие 139% больше медианы в 23 раза.
То есть относительно медианы, нашлось 52 контига с аномально большим покрытием
Выравнивание контигов на референсный геном
Как референс мы взяли Buchnera aphidicola str. W106 (Myzus persicae) strain W106 chromosome, complete genome NZ_CP002699.1
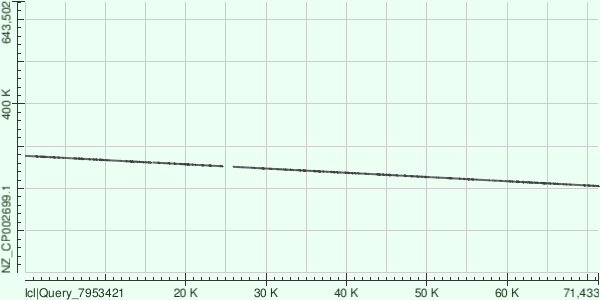
Контиг №11
Видим, что контиг ложится на довольно длинный участок хромосомы в начале и в конце. Это связано с тем, что начало секвенирования кольцевой ДНК референса попало ровно на наш контиг.
| № | Range | Score | Identities | Gaps |
|---|---|---|---|---|
| 1 | 628956 to 643122 | 12225 | 11780/14292(82%) | 272/14292(1%) |
| 2 | 36693 to 46298 | 11411 | 8484/9624(88%) | 49/9624(0%) |
| 3 | 1 to 11287 | 10532 | 9521/11390(84%) | 160/11390(1%) |
То есть первое по score выравнивание выровнялось к концу, далее идут выравнивания ближе к началу референсной последовательности.
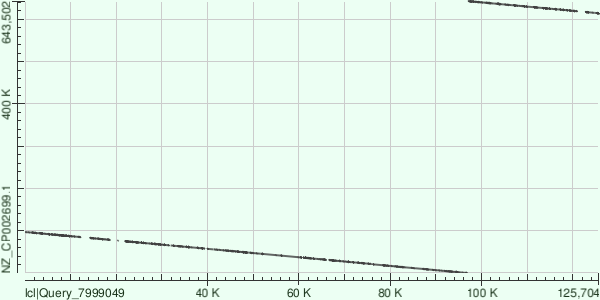
Контиг №1
Этот контиг на протяжении около 100Kb совпадвет с референсом.
| № | Range | Score | Identities | Gaps |
|---|---|---|---|---|
| 1 | 113933 to 140272 | 19826 | 21463/26651(81%) | 701/26651(2%) |
| 2 | 144142 to 166046 | 17407 | 18006/22172(81%) | 495/22172(2%) |
| 3 | 180920 to 199258 | 14916 | 15109/18531(82%) | 375/18531(2%) |
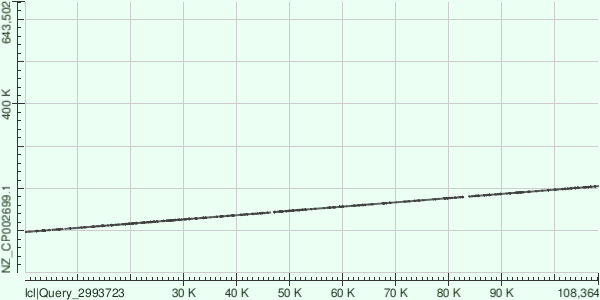
Контиг №14
Этот контиг как будто продолжает контиг №1, но на протяжении следующих 100Kb. Что интересно, он инвертирован. То есть правый конец ниже, чем левый.
| № | Range | Score | Identities | Gaps |
|---|---|---|---|---|
| 1 | 223109 to 235768 | 11457 | 10634/12789(83%) | 240/12789(1%) |
| 2 | 235791 to 248693 | 9640 | 10463/13024(80%) | 241/13024(1%) |
| 3 | 205664 to 216668 | 8826 | 9067/11149(81%) | 248/11149(2%) |