|
hhh
Hydrophobic
Hydrogen bonds
Disulfide bonds
Ligand
Resume
Save
|
|
hh
Ligand in general
Resume
Save
|
Primary characteristic of our structure
|
Antibodies are the group of glycoproteins that makes
up one of the main protein components of the blood
(approximately 20% of the total plasma protein by weight)
that have the ability to penetrate into the body's body fluids
and interact there with antigens.
In addition to antigen binding, antibodies have one or more effector functions.
The structural sites of the immunoglobulin molecule responsible for effector activity
(for the activation of a compliment or binding to phagocytic cells),
are spatially removed from the antigen-binding centers and they are mainly situate in the Fc region.
Our structure is a monoclonal mouse antibody in interaction with a hapten.
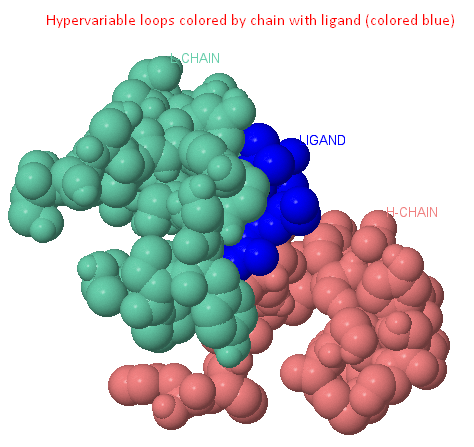
Hypervariable domains of antibodies
Each of the variable domains has its own set of three hypervariable regions stacked in three hypervariable loops.
Hypervariable loops of L- and H-variable domains are assembled into a group and form an antigen-binding site.
The variable region of the antibody molecule consists of conservative structure,
one of the ends of which are attached hypervariable loops (L1, L2, L3, H1, H2, H3 - shown in the last picture, Ligand button). [2][3][8]
|
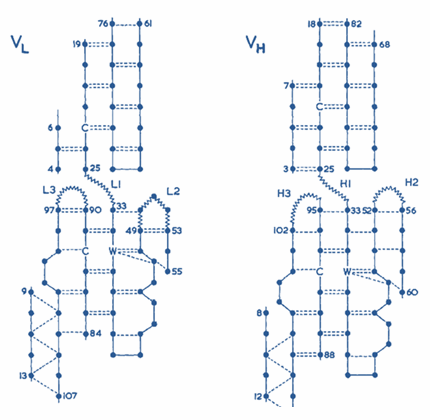
Pic. 1. Hypervariable domains of antibodies [2] |
Ligand description
General information about NPP ligand is represented in the table 1:
| PDB ID | -NPP |
| IUPAC | N-(2-AMINO-ETHYL)-4,6-DINITRO-N'-(2,2,6,6-TETRAMETHYL-1-OXY-PIPERIDIN-4-YL)-BENZENE-1,3-DIAMINE |
| Сhemical formula | 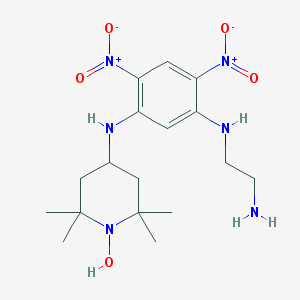 |
| Molecular formula | C17H28N6O5 |
| Molecular Weight | 396.448 g/mol |
| PubChem address | 444511 |
Intramolecular interactions
This section is about the research of intramolecular interactions. The two chains represented in the 1BAF structure may have following interactions:
1) Covalent (S-S cystein bridges)
2) Ionic (between amino acid side-chains with opposite full-electron charges)
3) Hydrogen bonds
4) Hydrophobic
Covalent interactions
Sulfur atoms colored belong to different chains. There is one disulfide bridge between chains and 4 intrachain bridges, two for each chain. Nevertheless, covalent bond is a very strong one and this disulfide bridge between chains must play its role in the protein folding.
Visualisation represented in the applet was received with the script.
Hydrogen bonds
Here, in different colors, different chains of structure are distinguished, darker colors indicate atoms that are not more than 3.5 angstrom from the opposite chain. Green color shows hydrogen bonds, there are 13 ones between chains.
 |
 |
| Domen | Chain | Number of hydrogen bonds in secondary structure | Number of alpha helixes | Number of beta structures |
|---|---|---|---|---|
| Domen 1 | L | 1 | 11 | 54 |
| Domen 2 | H | 1 | 10 | 24 |
| Domen 3 | H | 1 | 7 | 49 |
| Domen 4 | L | 2 | 8 | 58 |
The table gives a comparative characteristics of the elements of the secondary structure by chains and domains. The secondary structure of the protein is mainly represented by beta-strands, there are only 5 alpha-helices. Data is obtained with the help of the script.
Visualisation represented in the applet was received with the script.
Hydrophobic interactions
Here, in different colors, hydrophobic radicals of amino acids are marked, having phenylalanine (not more than 5 angstroms) in surrounding. The image in the applet shows 2 hydrophobic clusters consisting of side groups of amino acids of different chains. The boundaries of clusters can be determined more clearly, for instance, using the CluD service. However, since a large number of hydrophobic amino acids radicals of different chains face the opposite chain and form a cluster (in the applet distances between some atoms in clusters of hydrophobicity are signed), and without more detailed investigation it is possible to conclude, that hydrophobic interactions play an important role in the formation of the protein folding.
Visualization, available at the click of a button near the applet, was obtained with the help of the following script.
Ionic interactions
Balls of different colors show charged atoms having ionic interactions with atoms of the opposite chain. It can be stated that there are ionic interactions between the chains, but their number is very small. Since these bonds are weaker than all other types of studied interactions, we can conclude that their contribution to folding is insignificant.
| # | Atom with -NH3+-group | Atom with -COO--group | Distance between N-O atoms (A°) |
|---|---|---|---|
| Intrachain salt bridges | |||
| 1 | [LYS]13:H | [GLU]115:H | 3.80 |
| 2 | [ARG]39:H | [GLU]47:H | 3.25 |
| 3 | [ARG]39:H | [ASP]90:H | 2.89 |
| 4 | [ARG]60:L | [GLU]80:L | 3.54 |
| 5 | [ARG]60:L | [ASP]81:L | 4.00(OD1), 3.70(OD2) |
| 6 | [ARG]67:H | [ASP]90:H | 3.86 |
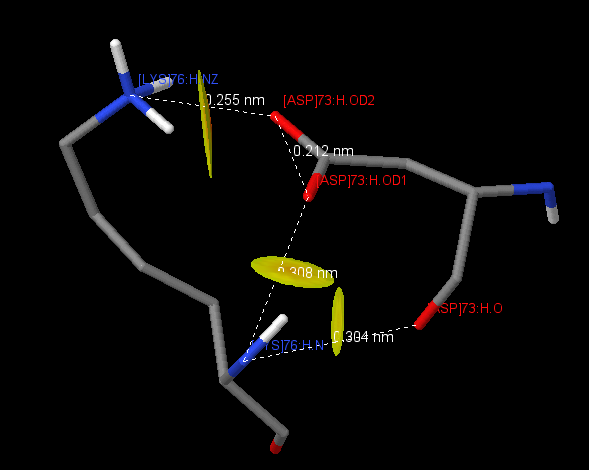
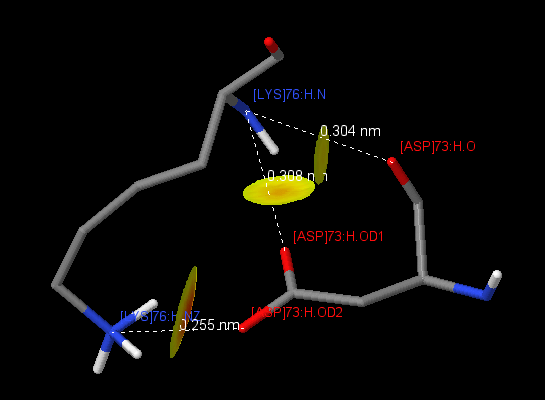
| 7 | [LYS]76:H | [ASP]73:H | 3.04(O), 3.08(OD1), 2.55(OD2) |
| 8 | [LYS]103:L | [GLU]105:L | 2.81 |
| 9 | [LYS]103:L | [ASP]165:L | 2.59(OD1), 2.74(OD2) |
| 10 | [LYS]142:L | [GLU]105:L | 2.80 |
| 11 | [LYS]199:L | [ASP]110:L | 2.74(OD1), 2.57(OD2) |
| 12 | [LYS]147:L | [GLU]154:L | 2.70(OE1)2.69(OE2) |
| 13 | [LYS]149:L | [GLU]195:L | 2.61(OE1), 2.70(OE2) |
| 14 | [LYS]183:L | [ASP]184:L | 2.65 |
| 15 | [ARG]188:L | [GLU]185:L | 3.94 |
| 16 | [ARG]215:H | [ASP]216:H | 3.94 |
| Interchain salt bridges | |||
| 1 | [LYS]210:H | [GLU]123:L | 2.75 |
| 2 | [ARG]215:H | [GLU]213:L | 3.11 |
Table 3 represents pairs of amino acid residues forming salt bridges. Salt bridges arise due to the attraction of two oppositely charged amino acids (Arg + , His + , Lys + , and Asp - , Glu - ), if the distance between the centers of charges is not more than 4 A [1] . These contacts were found using the script . The protein contains 16 intrachain salt bridges, 6 in the chain H and 10 in the chain L. 5 salt bridges of 16 contribute to parameters of the hydrogen bond (bond length 2.8-3.5 A). There is an interesting contact between [LYS] 76: H and [ASP] 73: H (Fig. 1), between which three salt bridges are formed at once. Only two interchain salt bridges were found, and both of them fit the definition of a hydrogen bond, from which it can be concluded that salt bridges do not make a significant contribution to the interaction between chains.
Visualization, available at the click of a button near the applet, was obtained with the help of the script. .
|
Description of hydrophobic core around Phe118L (core4)In the table below you can find information (hydrophobicity, heavy or light chain belonging) about amino acid residues situated on 1.0 - 7.0 A distance around Phe118. According to the CluD script, the centre of the core4 coincides with MyResidue (Phe118). About 60,5% of residues surrounding the Phe are hydrophobic (q.v. table). Distance between the residue and atoms that almost fully cover residue surface (100% Van Der Vaals radius) is more than 4 A. An interesting thing is that if the distance is enlarged to 5,6 A, “overshadowing” doesn’t increase notably. When the minimal distance between the Phe atoms and atoms of the surrounded residues is enlarged to 7 A, significant overshadowing of radical of MyResidue occurs. Characteristic average distance between neighboring not covalently bound atoms in the protein is near 4.1 (4.09) A.
If we neglect the radius of hydrogen atoms and assume the radius of the water molecule to be equal to the radius of oxygen, then it will fit between two covalently not bound atoms (if not a center of a hydrophobic nucleus is spoken, but the molecule in general). In hydrophobic cores (especially closer to its center), packing density of amino acid residues is higher and the distance between atoms is below the average (about 3.80 - 3.85 A). We can persume that water molecule can not fit the space between the atoms in the hydrophobic core center. In the last picture of the Hydrophobic section there is an image with the Phe colored according to the intensity of contacts with atoms of other residues. The greatest contribution to the interaction with the selected residue is made by Pro119, Ile117, Val133.
Process of hydrophobic core searcAt first, using restrict phe + wireframe 0.2 command, phenylalanines were chosen (according to workshop recommendations, as this amino acid has an aromatic hydrophobic radical with a relatively large molecular weight.) Next: select hydrophobic and not phe + wireframe 0.1, other hydrophobic amino acids were shown. Using define myresidue phe118:l Phe that visually seemed to be a center of the core was defined as MyResidue, and then considered residues of the environment using script refered above and the environment. The information can be found in the table.As mentioned earlier, the 1baf.pdb file was downloaded to CluD, from which a script to build hydrophobic cores in the protein molecule was obtained (more precisely, only some atoms of the amino acid residues that make up these nuclei were visualized). Amino acid residues from CluD were present in our sample a.k. core4. It is known that hydrophobic interactions are energetically favorable in aqueous solutions (in the absence of hydrophobic "tails", "overshadowing" water molecules from each other, more water molecules can form a greater variety of hydrogen bonds, and there is greater number of available "orientation options" as a result). In addition, hydrophobic interactions can facilitate establishment of several most favorable conformations of the protein molecule in aqueous solution. [1] |
Ligand-protein interactions
According to the results of the script there are no interactions between nitro groups and amino acid protein residues. Nitrogenes of the N-(2-amino-ethyl) group form neither salt bridges nor hydrogen bonds.
Piperidin ligand group is supposed to be a lipophilic region that can be approved by its surroundings: there are such amino acids as [TRP]90:L, [TRP]100:H and [TYR]31:L at the distance of 4.0 А from the group. Lipophilic ligand regions may account for penetration to the cell through the membrane lipid bilayer[5].
 |
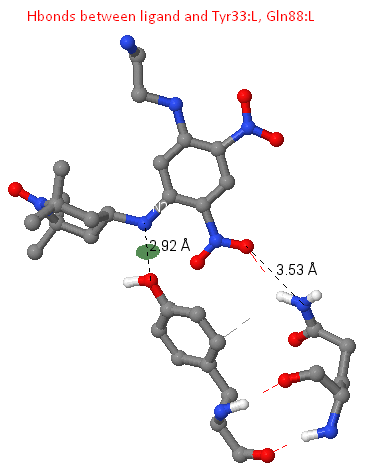 |
Ligand-protein hydrogen bonds
Between the N2 ligand atom and OH Tyr33: L, a hydrogen bond of length 2.92 A was found. There are data on the presence of so-called binding motifs (constant regions) in the composition of hypervariable loops of the antibody molecule. Such representer is the Tyr 32, chain L (the assumption was made on the basis of the fact that Tyr33: L participates in ligand binding in the structure we are considering, thus playing the role of the "lost" Tyr32: L, a kind of "compensation", the Tyr31: L is not involved in ligand binding). In the structure of the antibody we examined, this tyrosine is probably "shifted" to the 33 position of the L1 region.
In the structure under consideration, there is a hydrogen bond between the O2 atom of the ligand and the NE2 of the Gln88: L residue, having a length of 3.53 A (found using the calculate hbonds command).
Double pi-stacking
We should also mention the presence of double pi-stacking between the benzene ring of the ligand and the aromatic system of the amino acid residues of the antibody. Pi-stacking is a non-covalent interaction between organic compounds containing aromatic components, as the result of which pi-orbitals intersect, providing a gain in entropy of formed complex in aquatic environment. Pi-stacking is characterized by parallel arrangement of two aromatic systems at the distance of 3.5 - 4 A from each other. It should also be noted that the aromatic residues participating in pi-stacking are in most cases tryptophan and tyrosine. In the case of our structure, double pi-stacking is provided by the interaction of the ligand with Trp90: L (average distance is 3.94 A) and Trp100: H (mean distance is 3.52 A); (see pictures). The distance between the aromatic systems in the antigen-antibody complex under consideration corresponds to the optimal (according to the data of [4]). Thus, we can conclude that the described interaction is valid.
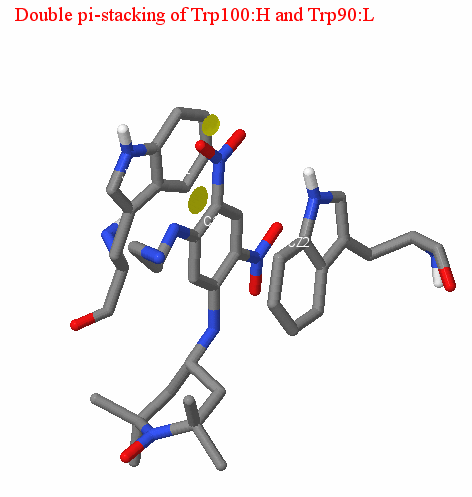 |
 |
Personal contribution
This project is executed by the excellent team of the fair sex: Belyaeva Julia, Nefedova Anastasia, Rosina Anna. BJ studied hydrophobic nuclei, ligand-protein contacts (pi-stacking, hydrogen bonds), and also took responsibility for loading applets and translation into Tatar. NA worked on ligand-protein interactions, salt bridges, intra-chain hydrogen bonds, in cooperation with RA translated into English. RA investigated all types of intermolecular contacts, disulfide bridges, made a general description of the protein, together with NA carried out the translation into English.
Literature list
[1] Tanford C. Contribution of hydrophobic interactions to the stability of the globular conformation of proteins //Journal of the American Chemical Society. – 1962. – n. 84. – no. 22. – p. 4240-4247
[2] Chothia C., Lesk A. M. Canonical structures for the hypervariable regions of immunoglobu-lins //Journal of molecular biology. – 1987. – n. 196. – no. 4. – p. 901-917
[3] North B., Lehmann A., Dunbrack R. L. A new clustering of antibody CDR loop conformations //Journal of molecular biology. – 2011. – n. 406. – no. 2. – p. 228-256
[4] Arzhanik V. et al. Interaction of antibodies with aromatic ligands: the role of pi-stacking //Journal of bioinformatics and computational biology. – 2010. – n. 8. – no. 03. – p. 471-483
[5] https://www.rcsb.org/structure/5LBY
[6] K.M. Zelenin: Physiologically active complexes of hydrazones, 1996
[7] https://www.ncbi.nlm.nih.gov/Structure/MMDB/docs/mmdb_help.html#WhatIs
[8] Martin A. C. R., Thornton J. M. Structural families in loops of homologous proteins: auto-matic classification, modelling and application to antibodies //Journal of molecular biology. – 1996. – n. 263. – ?. 5. – p. 800-815