Ресеквенирование. Поиск полиморфизмов у человека
Часть I. Анализ качества чтений. Очистка чтений
Все файлы, обозначенные в таблицах, можно посмотреть в рабочей директории,
указанной в задании
| Входной файл | Получаемый файл | Команда | Что делает |
| chr8.fastqc | chr8_fastqc.zip | fastqc chr8.fastq |
Контроль качества прочтений |
| chr8.fastq | out1.fastq | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr8.fastq out1.fastq TRAILING:20 |
Удаляем с конца нуклеотиды качеством < 20 |
| out1.fastq | out2.fastq | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 out1.fastq out2.fastq MINLEN:50 |
Удаляем с конца последовательности длины < 50 | out2.fastq | out2_fastq.zip | fastqc out2.fastq |
Контроль качества прочтений |
Per base sequence quality (до и после чистки)
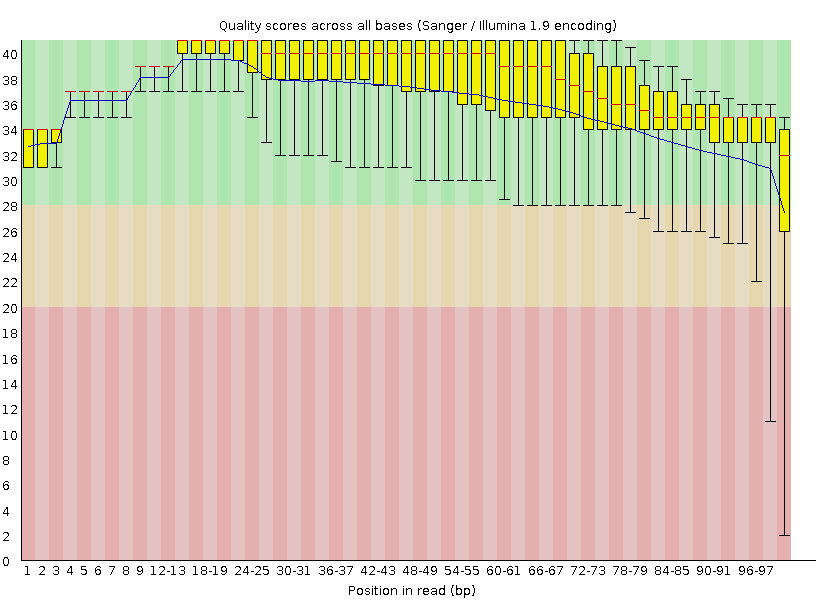 |
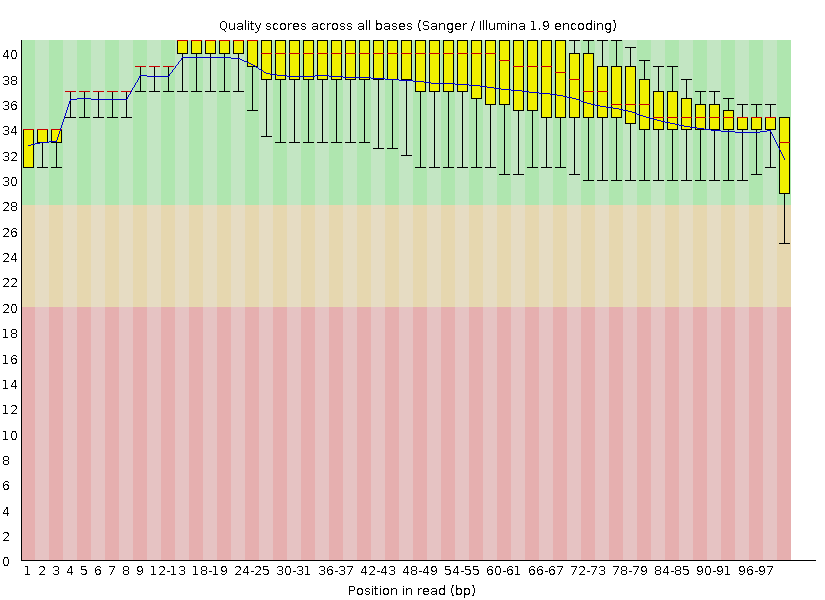 |
Число чтений до чистки: 8367
Число чтений после чистки: 8227
С помощью программы trimmomatic были удалены нуклеотиды с качеством чтения
(меньше 20, где Q=10*lg(p), p - вероятность ошибки)
Отобраны прочтения с длиной более 50 нуклеотидов
Пропали риды с длинными усами
В результате проведенной нами небольшой чистки количество прочтений уменьшилось на 140
Стоит обратить внимание на такой параметр как Per sequence GC content;
содержание GC пар изменилось с 39 до 38 %, тем не менее, до "чистки" рядом с этим параметром стояло
предупреждение (имеются отклонения от нормального распределения содержания GC-пар в более чем 15% прочтений),
после trimmomatic процент отклонения по прочтениям уменьшился, предупреждение пропало
Вероятно, предупреждение было связано с наличием коротких прочтений, которые вносили погрешности в подсчет
распределения процентного содержания GC-пар
Мы также можем заметить предупреждение напротив параметра Per base sequence content
Они возникает, если различие между А и Т, либо G и C больше 10% в какой-либо позиции
Это может быть связано с ошибкой в процессе секвенирования; скорее всего, это не опосредовано
присутствием какой-либо "загрязняющей" последовательности, иначе такие неточности систематически
появлялись бы в нескольких позициях, а не только в одной, как в нашем случае
Per base sequence content (до и после чистки)
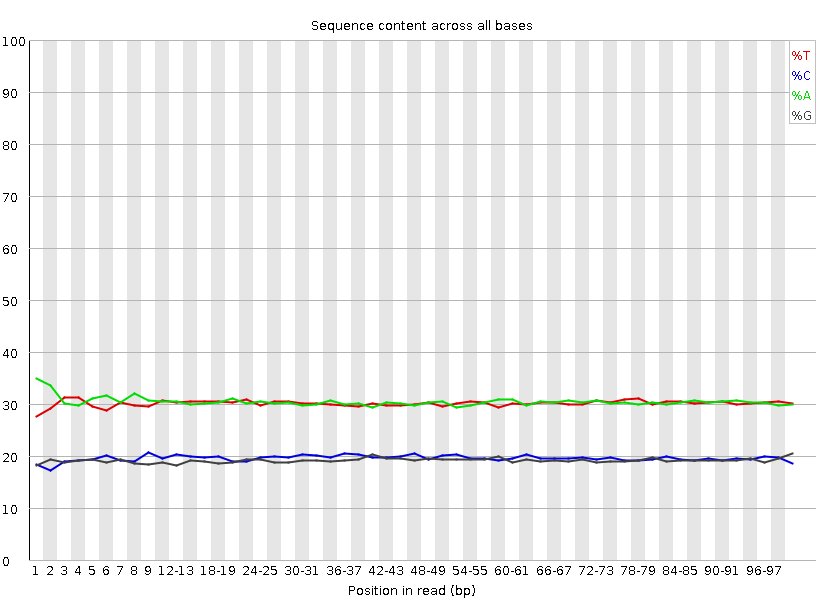 |
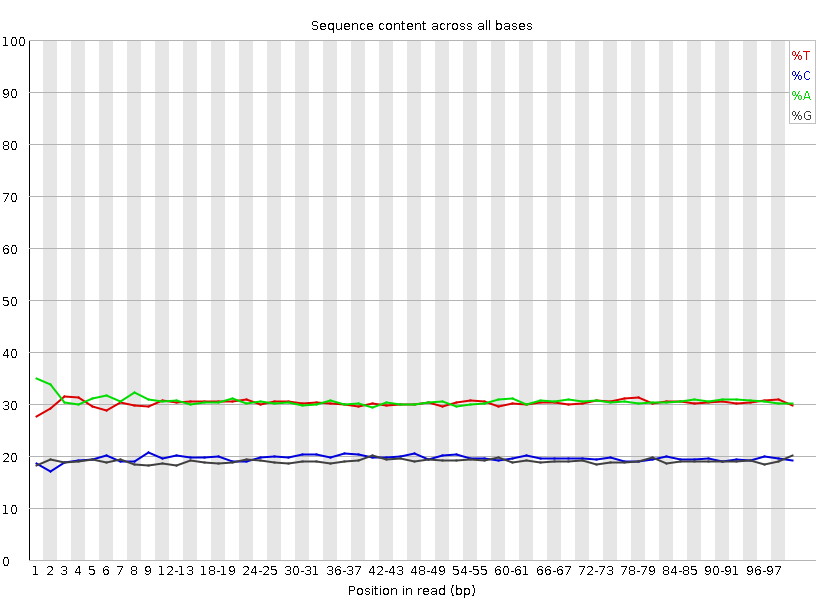 |
Sequence Length Distribution
Этот модуль генерирует график, показывающий распределение размеров ридов в анализируемом файле
Предупреждение возникает, если все прочтения имеют различную длину
В нашем случае длина ридов в большинстве случаев превышает 100 bp;
Не думаю, что в случае нашего файла такое предупреждение является серьезной проблемой, так как
данный параметр достаточно сильно зависит от качества секвенатора; так, если прибор не является
высокопроизводительным, то небольшие различия в длине ридов для него являются нормой
Sequence Length Distribution (до и после чистки)
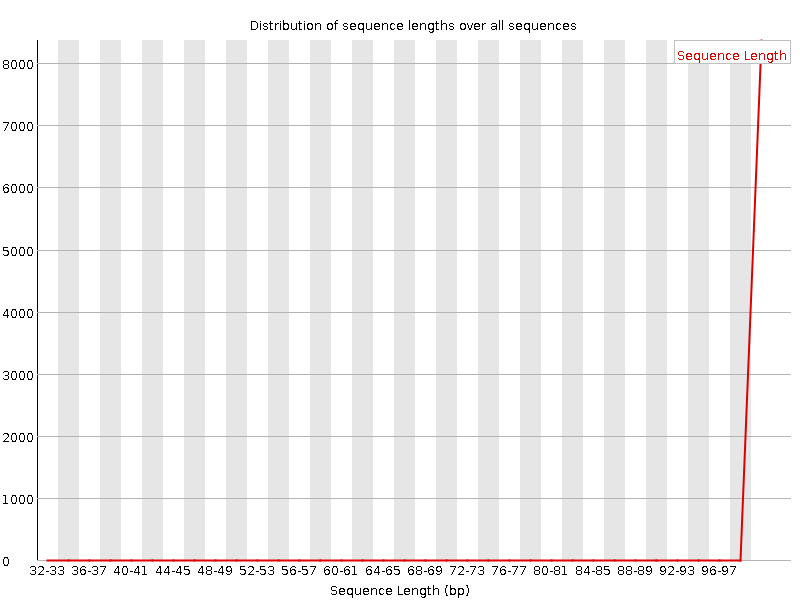 |
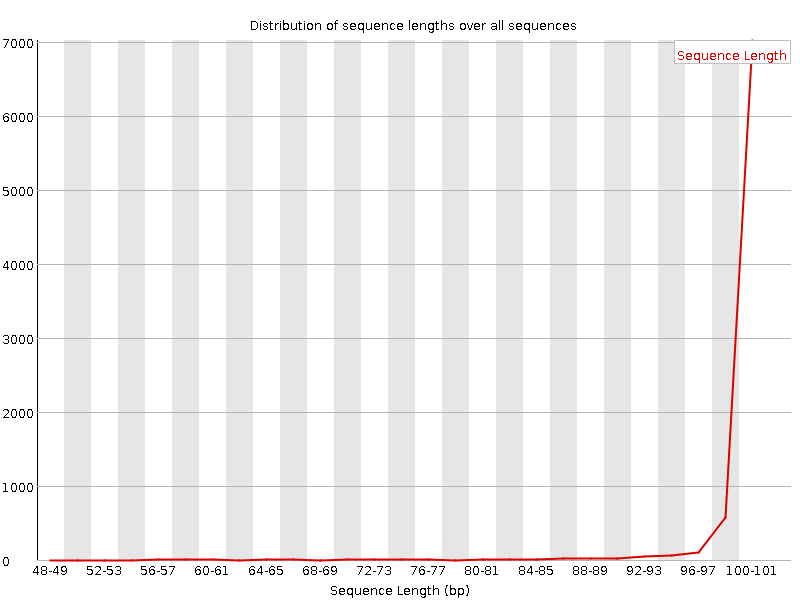 |
Часть II. Картирование чтений
| Входной файл | Получаемый файл | Команда | Что делает |
| chr8.fasta | mychr.* | hisat2-build chr8.fasta mychr |
Индексируем референсную последовательность |
| out2.fastq | samchr8.sam | hisat2 -x mychr -U out2.fastq -S samchr8.sam --no-spliced-alignment --no-softclip |
Строим выравнивание ридов и референса |
| samchr8.sam | bamchr8.bam | samtools view samchr8.sam -b >> bamchr8.bam |
Переводим выравнивание ридов с референсом в бинарный формат |
| bamchr8.bam | sortedbam.bam | samtools sort bamchr8.bam sortedbam |
Сортируем выравнивание ридов с референсом по координате в референсе начала чтения |
| sortedbam.bam | sortedbam.bam.bai | samtools index sortedbam.bam |
Индексируем отсортированный .bam файл |
Рассмотрим выдачу программы hisat2 - на геном откартировано 3227 ридов,
100% которых были непарными
8187 ридов (99.64%) выровнены ровно 1 раз
30 ридов (0.36%) выровнены 0 раз
0 ридов (0%) выровнены более 1 раза
С помощью команды
samtools depth sortedbam.bam >> cover.csvвычислим покрытие для каждого
нуклеотида. На выходе получаем таблицу; импортируем ее в Excel и построим гистограмму
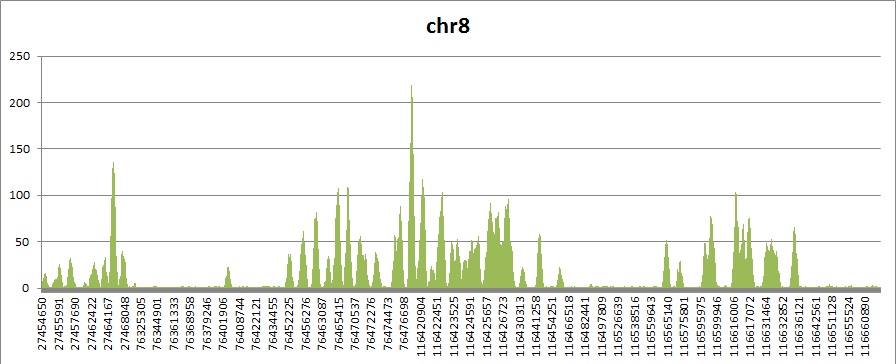
Наиболее высокие пики показывают нуклеотиды с самым высоким покрытием. Выберем один из них - 76 478 838
С помощью GenomeBrowser получаем информацию об интересующем нас нуклеотиде:
Homo sapiens hepatocyte nuclear factor 4, gamma (HNF4G), mRNA
hg19 chr8: 76 320 271 - 76 479 061
Размер: 158 791
Общее число экзонов: 11
Цепь: +
chr8: q21.11
Файл с последовательностями экзонов доступен по ссылке
Координаты экзона: 76 476 210 - 76 479 061 (наш нуклеотид - 76 478 838)
Запускаем команду
samtools depth -r chr8:76476210-76479061 sortedbam.bam >> exon_cover.csv
Для получения информации о покрытии нашего экзона. Полученную таблицу импортируем в Excel, находим
среднее покрытие экзона
Среднее покрытие экзона: 63.99
Таблица с информацией о покрытии экзона доступна по ссылке
Часть III. Анализ SNP
| sortedbam.bam, chr8.fasta | polymorphs.bcf | samtools mpileup -uf chr8.fasta -g -o polymorphs.bcf sortedbam.bam |
Создаем файл с полиморфизмами в .bcf |
| polymorphs.bcf | refandreaddiff.vcf | bcftools call -cv -o refandreaddiff.vcf polymorphs.bcf |
Создаем файл со списком отличий между референсом и ридами в .vcf |
Описание трех полиморфизмов из файла *.vcf
| Координата | Тип полиморфизма | В референсе | В ридах | Глубина покрытия | Качество чтения |
| chr8:27454785 | Индель | TAATGAA | TAA | 5 | 58.4663 |
| chr8:116599199 | Замена | T | G | 45 | 221.999 |
| chr8:76468309 (->76468321) | Индель | GTTTTTTTTTTTT | GTTTTTTTTTT,GTTTTTTTTT | 74 | 120.457 |
Индель chr8:27454785
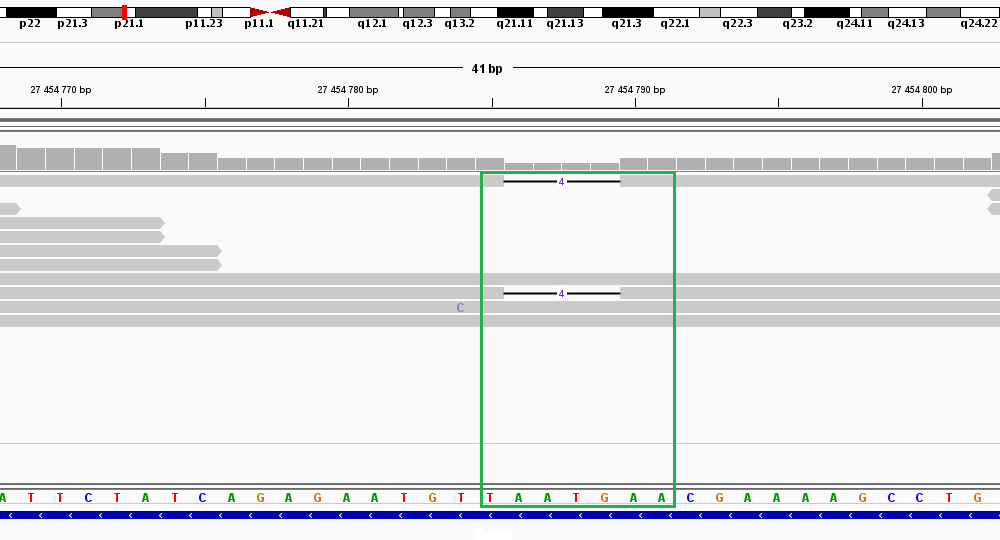
Замена chr8:116599199

Индель chr8:76468309

Обнаружено 100 полиморфизмов, в числе которых 5 инделей; 95 - замены (64 transitions and 31 transversions)
Среднее значение качества - 66.717
Среднее покрытие - 14.494
Глубина покрытия достаточно сильно варьируется для каждого из участков, при этом
среднее значение получается не слишком высоким
Следующий этап нашей работы - аннотация SNP
Нам необходимо проаннотировать полученные snp по 5 базам данных - refgene, dbsnp, 1000 genomes, GWAS, Clinvar
convert2annovar.pl -format vcf4 refandreaddiff.vcf > chr8.avinput |
Переводим файл .vcf формат, удобный для работы annovar |
annotate_variation.pl -filter -out SR_SNP -build hg19 -dbtype snp138 chr8.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Dbsnp |
annotate_variation.pl -out refgen -build hg19 chr8.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Refgene |
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000Genomes chr8.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по 1000 Genomes |
annotate_variation.pl -regionanno -build hg19 -out GWAS -dbtype gwasCatalog chr8.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Gwas |
annotate_variation.pl chr8.avinput -filter -dbtype clinvar_20150629 -buildver hg19 -out CLINVAR /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Clinvar |
При аннотации SNP по Refgen получаем три файла - refgen.exonic_variant_function, refgen.log
(системные сообщения, информация об ошибках и комментарии), refgen.variant_function
Из файла refgen.variant_function мы понимаем, что база данных Refseq делит snp по
их локализации (exonic - 5, intergenic - 17, intronic - 60, UTR3 - 13)
Гены, в которые попали наши snp:
CASC9 17 CLU 10 HNF4G 22 (+17) TRPS1 46
Помимимо всего прочего в файлах есть информация о синониминости (несинонимичности) замен
аминокислотных остатков
У 77 есть rs
Аннотация 1000Genomes показывает частоты аллелей в output - файле
Среднее значение частоты аллелей - 17,366 (подсчитано с помощью эксорта данных в Excel)
Также, в output - файле есть информация о характере замены (что на что было заменено - в форме гэпов и/или букв-нуклеотидов)
Также указаны координаты каждой такой замены
По аннотации Gwas видим, что 4 snp связаны с каким-либо заболеванием, либо физиологическим параметром
связь геномных признаков с фенотипическими:
Болезнь Альцгеймера (2 snp) 27456253 27456253 T C 3
27466315 27466315 T C 29
ЛВП (Липопротеины высокой плотности) 76478768 76478768 C T 134
Уровень мочевой кислоты 116599199 116599199 T G 45
ClinVar объединяет информацию о геномных вариациях (полиморфизмах), их отношении к здоровью человека В базе данных ClinVar не было какой-либо клинической информации по интересующим нас полиморфизмам
Полную таблицу вы можете загрузить cсылке