Часть I. Анализ качества чтений. Очистка чтений
| Входной файл |
Получаемый файл |
Команда |
Что делает |
| chr8.fastqc |
chr8_fastqc.zip |
fastqc chr8.fastq |
Контроль качества прочтений |
| chr8.fastq |
out1.fastq |
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar
SE -phred33 chr8.fastq out1.fastq TRAILING:20 |
Удаляем с конца нуклеотиды качеством < 20 |
| out1.fastq |
out2.fastq |
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar
SE -phred33 out1.fastq out2.fastq MINLEN:50 |
Удаляем с конца последовательности длины < 50 |
out2.fastq |
out2_fastq.zip |
fastqc out2.fastq |
Контроль качества прочтений |
Per base sequence quality (до и после чистки)
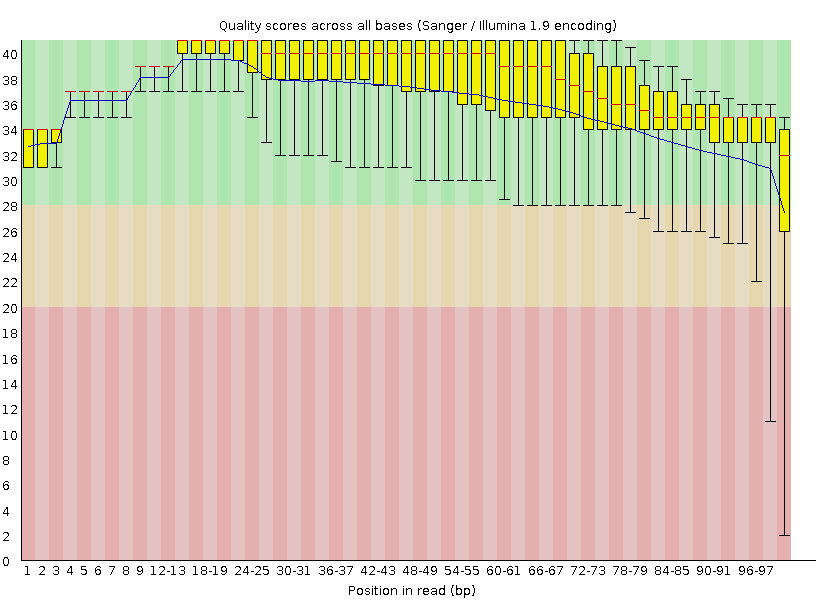 |
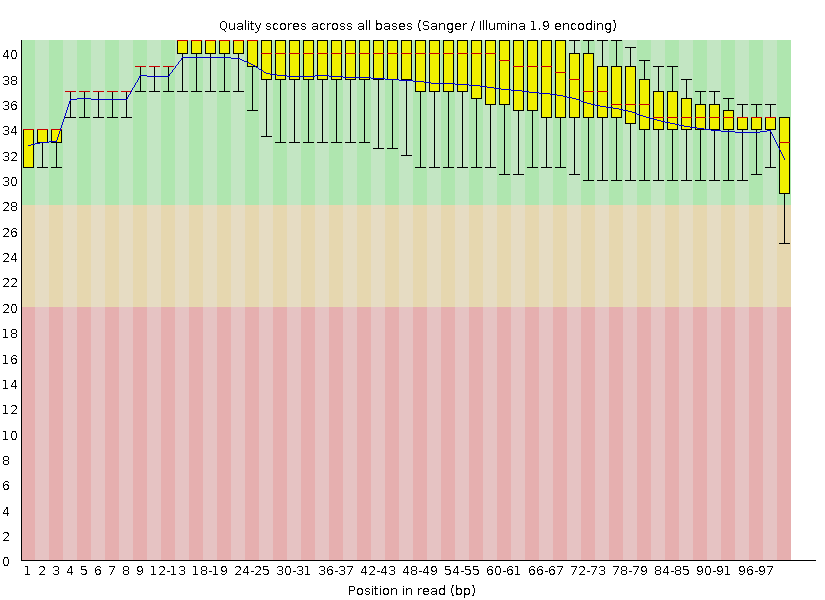 |
Число чтений до чистки: 8367
Число чтений после чистки: 8227
С помощью программы trimmomatic были удалены нуклеотиды с качеством чтения
(меньше 20, где Q=10*lg(p), p - вероятность ошибки)
Отобраны прочтения с длиной более 50 нуклеотидов
Пропали риды с длинными усами
В результате проведенной нами небольшой чистки количество прочтений уменьшилось на 140
Стоит обратить внимание на такой параметр как Per sequence GC content;
содержание GC пар изменилось с 39 до 38 %, тем не менее, до "чистки" рядом с этим параметром стояло
предупреждение (имеются отклонения от нормального распределения содержания GC-пар в более чем 15% прочтений),
после trimmomatic процент отклонения по прочтениям уменьшился, предупреждение пропало
Вероятно, предупреждение было связано с наличием коротких прочтений, которые вносили погрешности в подсчет
распределения процентного содержания GC-пар
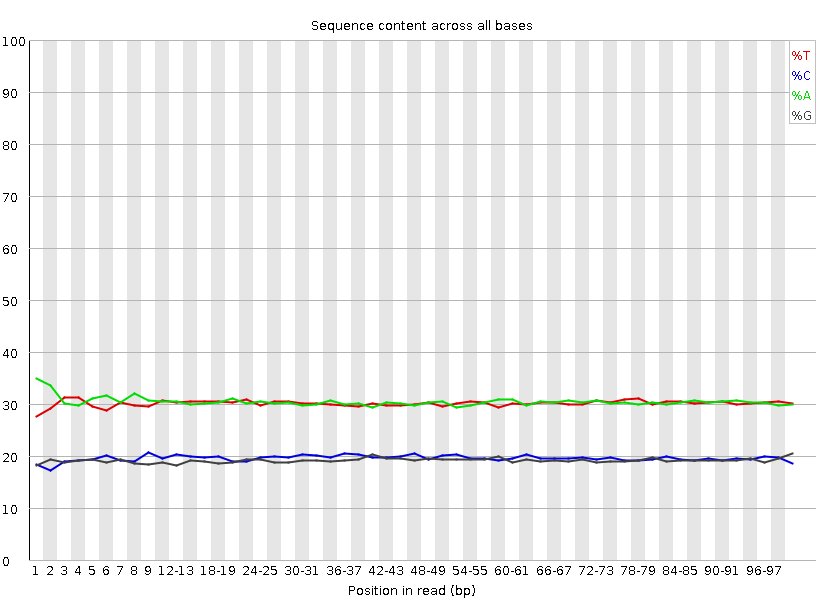
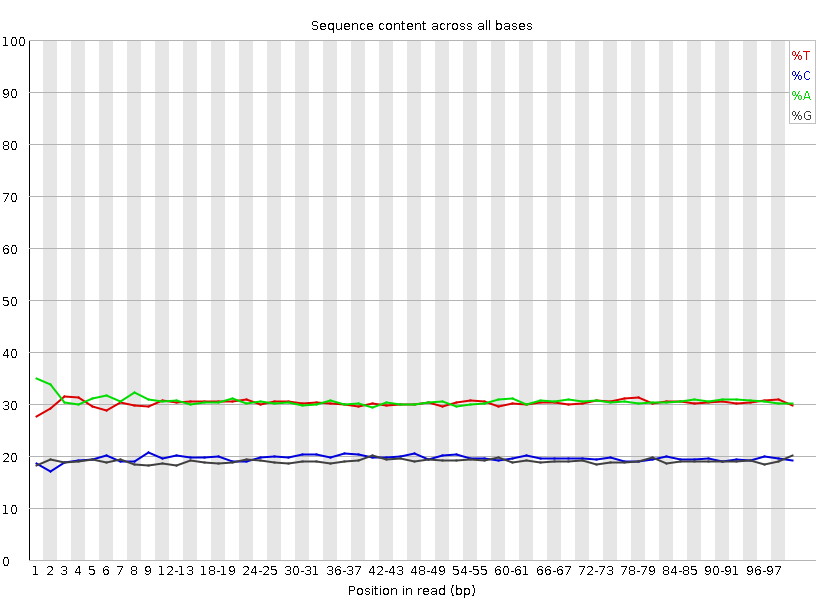
Мы также можем заметить предупреждение напротив параметра Per base sequence content
Они возникает, если различие между А и Т, либо G и C больше 10% в какой-либо позиции
Это может быть связано с ошибкой в процессе секвенирования; скорее всего, это не опосредовано
присутствием какой-либо "загрязняющей" последовательности, иначе такие неточности систематически
появлялись бы в нескольких позициях, а не только в одной, как в нашем случае
Per base sequence content (до и после чистки)
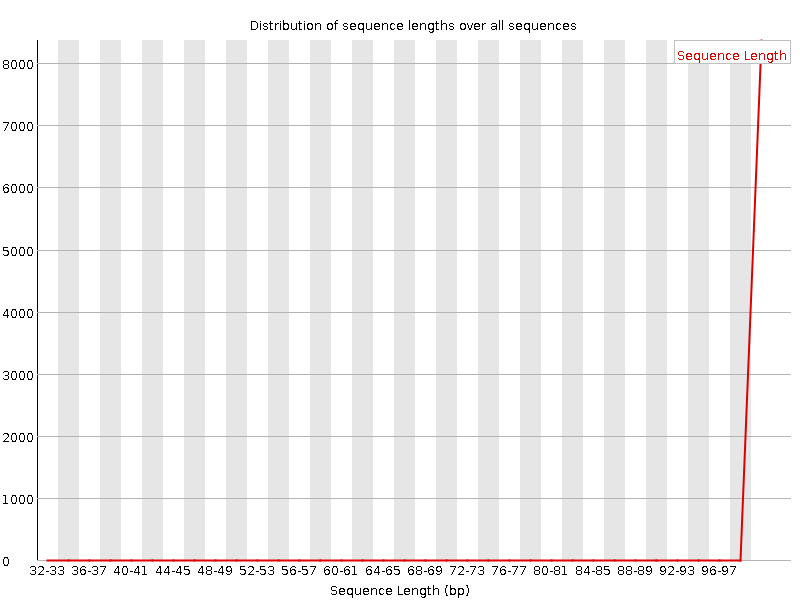
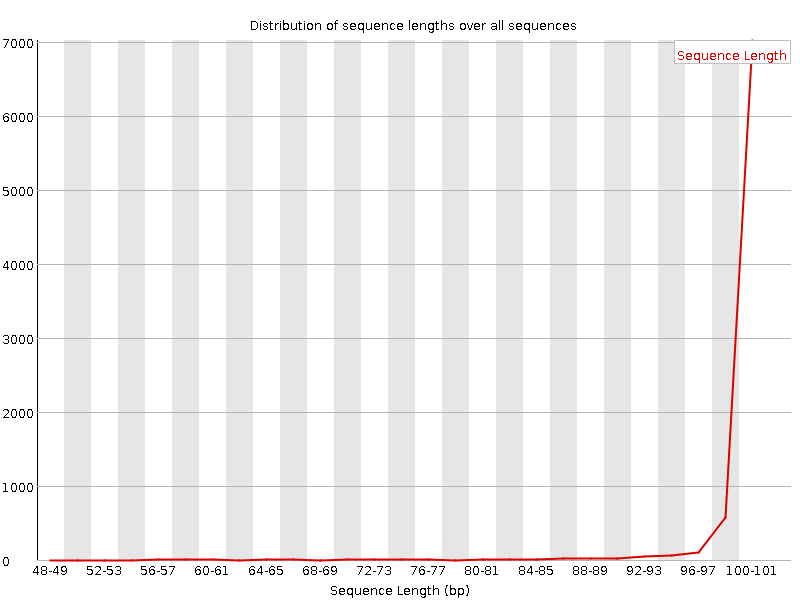
Sequence Length Distribution
Этот модуль генерирует график, показывающий распределение размеров ридов в анализируемом файле
Предупреждение возникает, если все прочтения имеют различную длину
В нашем случае длина ридов в большинстве случаев превышает 100 bp;
Не думаю, что в случае нашего файла такое предупреждение является серьезной проблемой, так как
данный параметр достаточно сильно зависит от качества секвенатора; так, если прибор не является
высокопроизводительным, то небольшие различия в длине ридов для него являются нормой
Sequence Length Distribution (до и после чистки)
Часть II. Картирование чтений
| Входной файл |
Получаемый файл |
Команда |
Что делает |
| chr8.fasta |
mychr.* |
hisat2-build chr8.fasta mychr |
Индексируем референсную последовательность |
| out2.fastq |
samchr8.sam |
hisat2 -x mychr -U out2.fastq -S samchr8.sam --no-spliced-alignment --no-softclip |
Строим выравнивание ридов и референса |
| samchr8.sam |
bamchr8.bam |
samtools view samchr8.sam -b >> bamchr8.bam |
Переводим выравнивание ридов с референсом в бинарный формат |
| bamchr8.bam |
sortedbam.bam |
samtools sort bamchr8.bam sortedbam |
Сортируем выравнивание ридов с референсом по координате в референсе начала чтения |
| sortedbam.bam |
sortedbam.bam.bai |
samtools index sortedbam.bam |
Индексируем отсортированный .bam файл |
Рассмотрим выдачу программы hisat2 - на геном откартировано 3227 ридов,
100% которых были непарными
8187 ридов (99.64%) выровнены ровно 1 раз
30 ридов (0.36%) выровнены 0 раз
0 ридов (0%) выровнены более 1 раза
С помощью команды
samtools depth sortedbam.bam >> cover.csv
вычислим покрытие для каждого
нуклеотида. На выходе получаем таблицу; импортируем ее в Excel и построим гистограмму
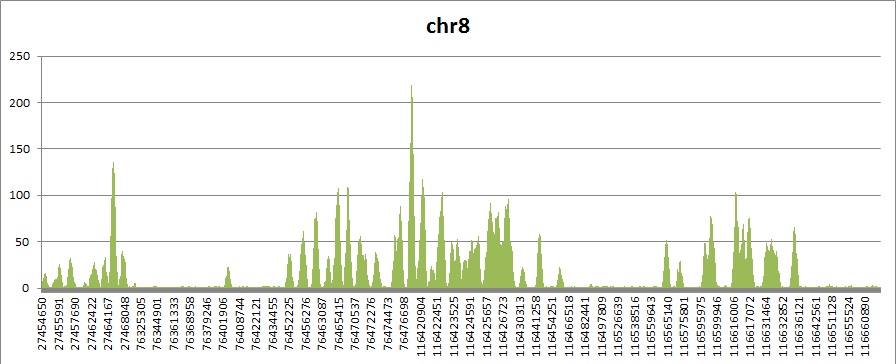
Наиболее высокие пики показывают нуклеотиды с самым высоким покрытием. Выберем один из них - 76 478 838
С помощью GenomeBrowser получаем информацию об интересующем нас нуклеотиде:
Homo sapiens hepatocyte nuclear factor 4, gamma (HNF4G), mRNA
hg19 chr8: 76 320 271 - 76 479 061
Размер: 158 791
Общее число экзонов: 11
Цепь: +
chr8: q21.11
Файл с последовательностями экзонов доступен по ссылке
Координаты экзона: 76 476 210 - 76 479 061 (наш нуклеотид - 76 478 838)
Запускаем команду samtools depth -r chr8:76476210-76479061 sortedbam.bam >> exon_cover.csv
Для получения информации о покрытии нашего экзона. Полученную таблицу импортируем в Excel, находим
среднее покрытие экзона
Среднее покрытие экзона: 63.99
Таблица с информацией о покрытии экзона доступна по ссылке