BLAST
Задание 1. Таксономия и функция прочтённой нуклеотидной последовательности (из практикума 6)
С помощью алгоритма blastn был произведен поиск гомологов консенсусной последовательности из практикума 6:
>EMBOSS_001 ttntnaaangacggccagtatggctcgtaccaagcagacngcacgtaaatYYWMCSGKKG RANGGCACCGCGAAAACAACTGGCCACCAAGGCAGCCCGMAAGAGTGCNCCAGCTACCGG NGGAGTGAAGAAACCTCATCGTTACAGGCCCGGGACNGTCGCTCTCCGTGAGATCCGTCG CTACCAGAAGAGCACCGAGCTCCTGATCCGAAAANTGCCCTTCCAGCGTCTGGTCAGAGA AATCGCYCAGGACTTCAAGACCGAGCTGCGNTTCCAGAGNTCCGCCNTCATGGCNCTCCA GGAAGCNAGCGAAGCCTACCTCGTCGGTCTCTTCGAGGACACCAACCTNTGCNccatyca cgccaaacgtgtcacnatnatgccnaaggatatgtcatagctgtttcn
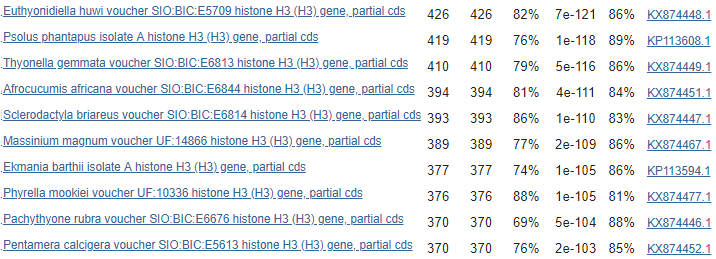
Приведу фрагмент таблицы находок (первые 6):
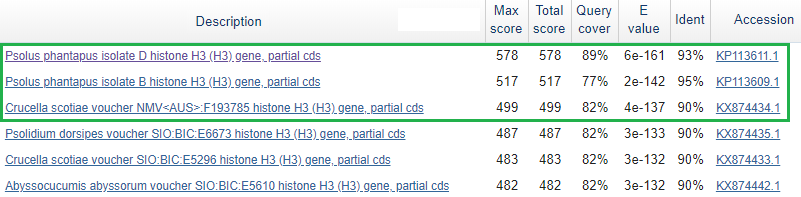
Начиная с четвертой находки значение e-value начинает убывать медленнее
(0-1 порядок на нашем фрагменте таблицы); рассмотрим первые три строки
Важно отметить, что во всех находках указан фрагмент гистона H3
В первых двух случаях совпадает и название организма - Psolus phantapus
Предположим, что участок нашей последовательности принадлежит именно ему (достаточно хорошие значения
e-value (6e-161 и 2e-142) и процента идентичности (93% и 95%)
Проверим наше предположение
Для этого рассмотрим парные выравнивания последовательностей первых трех находок
(в таксономии курсивом отметим таксоны, совпадающие у всех трех находок)
1) Psolus phantapus isolate D histone H3 (H3) gene, partial cds
E-value: 6e-161
Cover: 89%
Gaps: 0/365
Taxonomy:
Eukaryota; Metazoa; Echinodermata; Eleutherozoa; Echinozoa;
Holothuroidea; Dendrochirotacea; Dendrochirotida; Psolidae; Psolus
2) Psolus phantapus isolate B histone H3 (H3) gene, partial cds
E-value: 2e-142
Cover: 77%
Gap: 0/316
Taxonomy:
Eukaryota; Metazoa; Echinodermata; Eleutherozoa; Echinozoa;
Holothuroidea; Dendrochirotacea; Dendrochirotida; Psolidae; Psolus
3) Crucella scotiae voucher NMV:F193785 histone H3 (H3) gene, partial cds
E-value: 4e-137
Cover: 82%
Gaps: 0/335
Taxonomy:
Eukaryota; Metazoa; Echinodermata; Eleutherozoa; Echinozoa;
Holothuroidea; Dendrochirotacea; Dendrochirotida; Paracucumidae
Постараемся определить семейство. Построим множественное выравнивание из четырех последовательностей
В последовательности Crucella scotiae имеются замены в 70 и 73 позициях
в сравнении с другими последовательностями:

Также можно привести в пример замены в позициях 181 и 214 той же находки
Замены в других находках более редкие. Таким образом, делаем вывод
о вероятной принадлежности нашего организма к семейству Psolidae, роду Psolus и виду Psolus phantapus

Также можно привести в пример дерево, построенное на основе выравнивания:

Для определения принадлежности последовательности из двух отобранных ранее находок выберем
ту, что имеет лучшее e-value; таким образом, в нашей последовательности находится фрагмент гена
гистона Н3 (как упоминалось ранее). Для простоты приведу фрагмент записи:
gene <1..>365
/gene="H3"
mRNA <1..>365
/gene="H3"
/product="histone H3"
CDS <1..>365
/gene="H3"
/codon_start=2
/product="histone H3"
/protein_id="AJS14690.1"
/translation="TASMARTKQTARKSTGGKAPRKQLATKAARKSAPATGGVKKPHR
YRPGTVALREIRRYQKSTELLIRKLPFQRLVREIAQDFKTELRFQSSAIMALQEASEA
YLVGLFEDTNLCAIHAKRV"
Интересна следующая строчка в записи банка: /country="Russia: Barents Sea"
Для итоговой проверки, мы можеv транслировать нашу последовательность из практикума 6 и просмотреть
результаты множественного выравнивания (все это - на основе алгоритма blastx). По полученным
находкам делаем вывод о том, что продукт нашего гена действительно гистон H3
Фотография Psolus phantapus

Задание 2. Сравнение списков находок нуклеотидных последовательностей тремя разными вариантами blast
Таблицу с данными по трем разным вариантам алгоритмов blast вы можете скачать по ссылке
| Алгоритм | Параметры алгоритма | Число находок | Комментарии |
| megablast | Стандартные; длина слова=28 | 4975 | Последняя находка с e-value=9e-33; несколько до нее - 2е-33; был выбран параметр "показать первые 5000 находок" |
| blastn | Стандартные; длина слова=11 | >5000 | Был выбран параметр "показать первые 5000 находок"; последняя находка на странице с e-value=4e-87 |
| blastn | Длина слова=7 | >5000 | Был выбран параметр "показать первые 5000 находок" |
Перейдем к таксономическим категориям для ограничения нашего поиска
Первоначально, при первом вызове алгоритмов, с использованием стандартным параметров, алгоритм megablast показал наименьшее
количество находок (4975 из 5000), что, вероятно, связано с длиной слова 28 (как следствие - выдача более близких гомологов).
Далее мы понижали ранг таксонов, фиксируя значения e-value первой и последней находки на странице, а также количество записей (из 100) на странице
Прослеживается общая тенденция по понижению e-value первой записи с понижением ранга таксона 1-3 порядка, что объяснимо все большим ограничением
выборки до более родственных организмов. Причем наиболее низкие значения e-value наблюдаются при использовании алгоритма blastn с параметрами, отличными от
стандартных - уменьшение длины слова до 7 и изменение значений параметра Match/Mismatch Scores до более чувствительных
Наибольшие значения e-value, хоть и с небольшой разницей с megablast показывает blastn со стандартными параметрами
В последнем вызове по наименьшему таксону e-value всех находок вновь повышается
В случае с e-value последних записей, напротив, мы наблюдаем постепенное увеличение e-value находок при понижении ранга таксонов,
ограничивающих поисковую базу, причем в данном случае наиболее высокие значения наблюдаются у алгоритма megablast
Последние два вызова характеризуются понижением e-value, что не соответствует неблюдаемой ранее танденции к понижению
Интересно, что при ограничении таксона до Paracucumidae e-value blastn с "нестандартными" параметрами имеет значение > 0
Возможно, это связано с использованием короткого поискового слова):

В случае с поиском по ограничению Paracucumidae megablast показал 2 находки, blastn с длиной слова 7 -
8 находок, причем подходящими по e-value были только 3 из них и 3 находки blastn со стандартными параметрами
Критическое понижение по количеству находок наблюдалось при ограничении поиска таксоном Dendrochirotacea, когда количество
результатов снизилось от >100 до 29 (для всех алгоритмов); это, вероятно, говорит о повышенной специфичности рассматриваемой нами
структуры внутри данного таксона
Приведу в пример выдачу алгоритмов, полученную на описанном выше этапе (ограничение на 50 находок, 20 - 29 находки):
megablast
blastn7
blastn11
Задание 3. Наличие гомологов белков в неаннотированном геноме
Amoeboaphelidium protococcarum
При запуске локального blast по алгоритму tblastn, query=HSP71_YEAST, outfmt: 7
Была получена следующая таблица:
query id subject id %ident aln len mis gap opens q. start q. end s. start s. end evalue bit score P10591|HSP71_YEAST scaffold-199 78.98 609 125 2 2 607 1109256 1107430 0.0 920 P10591|HSP71_YEAST scaffold-199 55.56 27 12 0 82 108 1110027 1109947 0.002 40.8 P10591|HSP71_YEAST scaffold-96 66.06 607 201 4 3 606 89928 91742 0.0 744 P10591|HSP71_YEAST scaffold-423 65.73 607 203 4 3 606 1313216 1311402 0.0 737 P10591|HSP71_YEAST scaffold-423 37.34 391 228 5 4 378 781726 782895 8e-64 232 P10591|HSP71_YEAST unplaced-999 81.90 315 56 1 2 315 945 1 8e-171 540 P10591|HSP71_YEAST unplaced-980 77.54 334 73 1 276 607 1 1002 9e-142 461 P10591|HSP71_YEAST scaffold-157 46.77 402 196 6 216 607 165338 166519 1e-81 285 P10591|HSP71_YEAST scaffold-157 54.42 215 93 3 5 217 164418 165053 5e-64 233 P10591|HSP71_YEAST scaffold-157 29.03 310 193 5 61 343 219904 218975 5e-32 133 P10591|HSP71_YEAST scaffold-693 46.77 402 196 6 216 607 1114528 1115709 2e-80 281 P10591|HSP71_YEAST scaffold-693 54.42 215 93 3 5 217 1113608 1114243 1e-63 232 P10591|HSP71_YEAST scaffold-693 27.79 331 212 5 61 364 1168679 1167687 2e-29 125 P10591|HSP71_YEAST unplaced-804 71.50 193 53 1 417 607 17964 17386 2e-74 264 P10591|HSP71_YEAST scaffold-499 71.50 193 53 1 417 607 3580 4158 6e-74 262 P10591|HSP71_YEAST unplaced-959 37.34 391 228 5 4 378 9193 10362 1e-63 231 P10591|HSP71_YEAST scaffold-469 76.92 104 24 0 437 540 2 313 5e-43 150 P10591|HSP71_YEAST scaffold-418 76.92 104 24 0 437 540 312 1 5e-43 150 P10591|HSP71_YEAST unplaced-113 68.97 87 26 1 9 94 1 261 1e-32 122 P10591|HSP71_YEAST scaffold-138 57.75 71 28 1 539 607 249 37 3e-17 78.6 P10591|HSP71_YEAST scaffold-61 57.75 71 28 1 539 607 5 217 3e-17 78.6 P10591|HSP71_YEAST unplaced-721 68.97 29 9 0 579 607 272 186 2e-05 43.9Наименьшего значения e-value достигают фрагменты scaffold-199 (609 ао), scaffold-96 (605 ао) и scaffold-493 (605) (0.0)
Интересно, что s-96 и s-493 имеют одинаковую длину, а s-199 отличается от них всего на 4 остатка (если транслировать в
аминокислотную последовательность. Длина query составляет 542 аминокислотных остатка, таким образом, покрытие query
составляет 94,9% и по 94,2%, что наряду с хорошим значением e-value позволяет предположить, что шаперон HSP70 имеет
положительные гомологи в геноме Amoeboaphelidium protococcarum
Для интереса произведем парное выравнивание участков s-199 и s-96, s-96 и s-493, гомологичных последовательности шаперона
При выравнивании s-199 и s-96 blastp показал 0.0 e-value при 64% идентичности, выравнивание s-96 и s-493 ожидаемо показало
e-value=0.0 и 99% идентичности (в одной из последовательностей остаток глутамина показан *)
Таким образом, мы можем предположить наличие двух положительных гомологов шаперона HSP70 в интересующем нас геноме, причем
один, вероятно, представлен в двух копиях в составе разных скэффолдов
При query=PRPC_EMENI получаем следующую выдачу:
query id subject id %ident aln len mis gap opens q. start q. end s. start s. end evalue bit score Q9TEM3|PRPC_EMENI scaffold-693 56.38 376 158 4 86 460 1243882 1244994 6e-121 393 Q9TEM3|PRPC_EMENI scaffold-693 41.79 67 38 1 20 85 1243614 1243814 5e-07 52.0 Q9TEM3|PRPC_EMENI scaffold-157 56.38 376 158 4 86 460 314582 315694 6e-120 390 Q9TEM3|PRPC_EMENI scaffold-157 40.30 67 39 1 20 85 314314 314514 8e-07 51.2 Q9TEM3|PRPC_EMENI scaffold-287 26.27 373 217 17 117 451 548001 546943 9e-11 64.3 Q9TEM3|PRPC_EMENI scaffold-212 27.82 266 163 12 199 453 46016 45273 1e-08 57.4Первые 4 находки представляют собой последовательности в составе двух скэффолдов - 693 и 157.
Более низкие значения e-value имеют последовательности, находящиеся внутри промежутка более крупных, с хорошим e-value
Вероятно, эти "концевые" участки важны для выявления более близкой гомологии
Посмотрим на покрытие интересующих нас находок (1 и 3 строки приведенной выше таблицы). Длина query составляет 460 ао, длина
участков гомологии 371 и 371. Покрытие 80,7% в обоих случаях, что, наряду со значениями e-value 6e-121 и 6e-120 позволяет предположить
наличие положительных гомологов. Заметим, что длины участков гомологии одинаковы, значения e-value отличаются на порядок
Проверим возможность того, что последовательности scaffold-693 и scaffold-157 одинаковы (в одной из них могут быть не "прочитаны" какие либо
остатки. Произведем парное выравнивание с применением алгоритма blastp, предварительно выгрузив гомологичные участки белковоц последовательности
из файла с выравниванием (проверка очень условная). Результаты такого локального выравнивания:
Query 1 GIRFRGMTIPEC*EKLPKANGG*EPLPEGLFYLLLTGEVPTKEQVDEVSRDWANRASSLP 60
GIRFRGMTIPEC*EKLPKANGG*EPLPEGLFYLLLTGEVPTKEQVDEVSRDWANRASSLP
Sbjct 1 GIRFRGMTIPEC*EKLPKANGG*EPLPEGLFYLLLTGEVPTKEQVDEVSRDWANRASSLP 60
Query 61 KHVEDIID*CPVTLHPMSQFSIAVTAMQHDSKFAQAYQQGVHKSKYWEYAYEDSMDLIAK 120
KHVEDIID*CPVTLHPMSQFSIAVTAMQHDSKFAQAY QGVHKSKYWEYAYEDSMDLIAK
Sbjct 61 KHVEDIID*CPVTLHPMSQFSIAVTAMQHDSKFAQAY*QGVHKSKYWEYAYEDSMDLIAK 120
Query 121 LPVVASRIYRNVFKDGKVAAIDKTKDWSYNFANMLGFGKDAQFVELMRLYLTIHSDHEGG 180
LPVVASRIYRNVFKDGKVAAIDKTKDWSYNFANMLGFGKDAQFVELMRLYLTIHSDHEGG
Sbjct 121 LPVVASRIYRNVFKDGKVAAIDKTKDWSYNFANMLGFGKDAQFVELMRLYLTIHSDHEGG 180
Query 181 NVSAHTTHLVGSALSDPYLSFAAGLNGLAGPLHGLANQEVLRWILQMKEEIGTNVSDEQV 240
NVSAHTTHLVGSALSDPYLSFAAGLNGLAGPLHGLANQEVLRWILQMKEEIGTNVSDEQV
Sbjct 181 NVSAHTTHLVGSALSDPYLSFAAGLNGLAGPLHGLANQEVLRWILQMKEEIGTNVSDEQV 240
Query 241 RDYCWKTLKSGQVIPGYGHAVLRKTDPRYTCQREFALKHLPTDPLFKMVSQLYNIVPNVL 300
RDYCWKTLKSGQVIPGYGHAVLRKTDPRYTCQREFALKHLPTDPLFKMVSQLYNIVPNVL
Sbjct 241 RDYCWKTLKSGQVIPGYGHAVLRKTDPRYTCQREFALKHLPTDPLFKMVSQLYNIVPNVL 300
Query 301 TEQGKTKNPFPNVDAHSGVLLQHYNLKEQEFYTVLFGVSRALGCLSQLVWDRALGLPIER 360
TEQGKTKNPFPNVDAHSGVLLQHYNLKEQEFYTVLFGVSRALGCLSQLVWDRALGLPIER
Sbjct 301 TEQGKTKNPFPNVDAHSGVLLQHYNLKEQEFYTVLFGVSRALGCLSQLVWDRALGLPIER 360
Query 361 PKSLTTDTIKK 371
PKSLTTDTIKK
Sbjct 361 PKSLTTDTIKK 371
Как мы и предполагали, Q обозначен как * в одно из последовательностей; гомология участков хорошая (e-value=0.0, cover=99%)
Таким образом, мы можем сделать вывод, что в геноме есть положительный гомолог митохондриальной цитратсинтазы в двух копиях
в составе разных скэффолдов
Третий вариант белка - TERT_SCHPO (query):
query id subject id %ident aln len mis gap opens q. start q. end s. start s. end evalue bit score O13339|TERT_SCHPO scaffold-17 25.05 491 305 16 320 780 610900 612273 1e-23 108 O13339|TERT_SCHPO scaffold-17 35.85 53 33 1 505 557 639070 639225 4.1 30.8 O13339|TERT_SCHPO unplaced-307 26.84 503 282 17 320 780 14863 16239 7e-22 102 O13339|TERT_SCHPO scaffold-105 28.26 92 62 1 810 897 24415 24140 0.51 33.9 O13339|TERT_SCHPO unplaced-647 41.67 36 21 0 170 205 141 248 4.9 28.1Из 5 вариантов находок 3, вероятно, не являются гомологами (если судить по значению e-velue (интересно, что вариант с e-value=4.1
входит в состав скэффолда 17, расширенный участок которого представляет собой наилучшую находку)
Обратим внимание на процент покрытия. Общая длина теломеразы (query) составляет 988 аминокислотных остатка; размеры участков гомологии
двух наилучших находок - 458 (46,4%) ао и 459 (46,5%). Таким образом, можно сделать вывод, что если последовательности и являются гомологами,
то условно положительный (недостаточно большое покрытие последовательности, используемой для выравнивания и высокие, относительно предыдущих
двух белков значения e-value; таким образом, нельзя точно говорить о сохранении функциональных и структурных свойств
Задание 4. Ген 40S-рибосомального белка S4 в scaffold-497
С помощью команды infoseq из пакета EMBOSS мы получили таблицу с именами и длинами скэффолдов
Для удобства испортируем полученную таблицу в Excel и отсортируем по убыванию длины. Для дальнейшей работы возьмем 51 строку таблицы -
scaffold-497 длины 70081. С помощью seqret получили нуклеотидную последовательность интересующего нас скэффолда и провели выравнивание
по алгоритму megablast с длиной слова=20 и Match/Mismatch Scores=[1,-2]. Получили следующую картину:
Лучшие находки предлагают примерно одинаковые рамки query-последовательности: 32165 - 32656 (32667; возьмем более "длинный" вариант); выберем этот
участок последовательности и построим выравнивание еще раз (уменьшим длину слова до 16)
Интересующий нас фрагмент скэффолда:
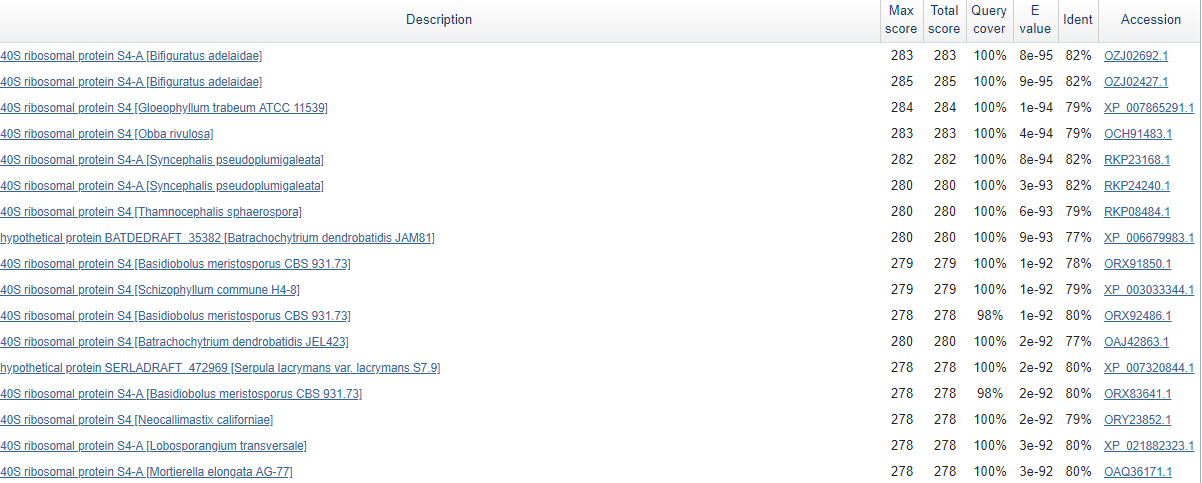
>scaffold-497_32165-32656 GATGTCATTTCCATTGAGAAGACTGGTGAGCACTTCCGTCTTGTGTATGATGTCAAGGGT CGCTTCACTATCCACCGTATCACTGATGAAGAGGCCAAGTTCAAGCTGTGCAAGGTTAGA AAGGTTCAGCTGGGAGCTAAAGGTATTCCATACGTTGTCACTCATGATGGCCGCACCATC CGTTACCCGGATCCATTGATCAAAGCTAATGACACTGTGAAGGTTGA-TCTG-GAGACTG GAAAGATTGTCGACTTTGTCAAGTTTGACACTGGTAACATGTGTGCCATCACTGGCGGTC GTAACATGGGTCGTATGGGTGTCATTGTGCACCGTGAGAGACATCTGGGTGGTTTCGATA TTGTCCACATCAAGGACGCGTTGGATCACACCTTCGCCACTCGTCTGAGCAACGTCTTTG TCATTGGTAAGGGTAACAAGGCTTGGGTGTCTCTGCCAAAGCAGAAGGGTGTCAAGCTGA CCATCCTGGAGGAGПолучаем следующие находки (участок таблицы):

Светло-зеленым отмечены находки рибосомального (40S) белка S4, темно-зеленым - мРНК гипотетических, неохарактеризованных белков
которые, вероятно, могут подходить интересующему нас множеству. E-value находок, процент покрытия достаточно хорошие
Таким образом, мы предполагаем, что нашли реально существующий ген 40S-рибосомального белка S4 с упомянутыми ранее границами 32165 - 32656
Сделаем еще одну проверку, построив выравнивание по алгоритму blastx со стандартными параметрами:
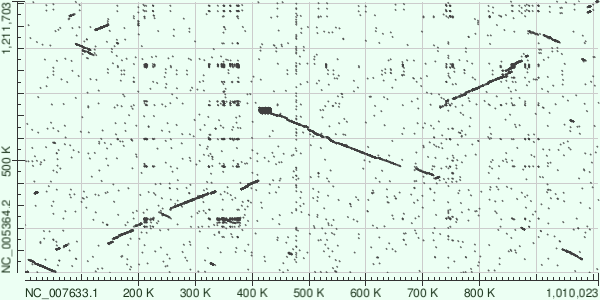
Задание 5. Карта локального сходства геномов двух бактерий
Для построения карты локального сходства выберем два генома -
Mycoplasma capricolum NC_007633.1 и Mycoplasma mycoides NC_005364.2
На карте можно заметить несколько инверсий - ~ 0-50К нуклеотидов (небольшая, в самом начале),
~ 410-670К (наиболее крупная), ~680-730К (небольшая), ~880-940К (небольшая в конце)
Также можно обратить внимание на крупную делецию (между 50 и 150К)
Помимо всего прочего, в конце карты (смотрим координаты, как и ранее, по оси Х) присутствует
небольшой инвертированный участок, смещенный почти к 0 (если смотреть по оси У),
причем при проекции концевых точек на ось У координаты нижней точки практически соответствуют
координатам верхней точки упомянотого выше (самым первым) инвертированного участка)
Возможно, что этот "конечный" участок был перемещен в одном из геномов (но это не точно)