
Все операции были выполнены с помощью данных команд:
fastqc chr9_1.fastq
команда выдает анализ качества чтений (2 файла: chr9_1_fastqc.html chr9_1_fastqc.zip)
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr9_1.fastq chr9_1_short.fastq SLIDINGWINDOW:50:20
Команда сканирует прочтения окном в 50, а потом удаляет с конца прочтения куски качество которых ниже 20
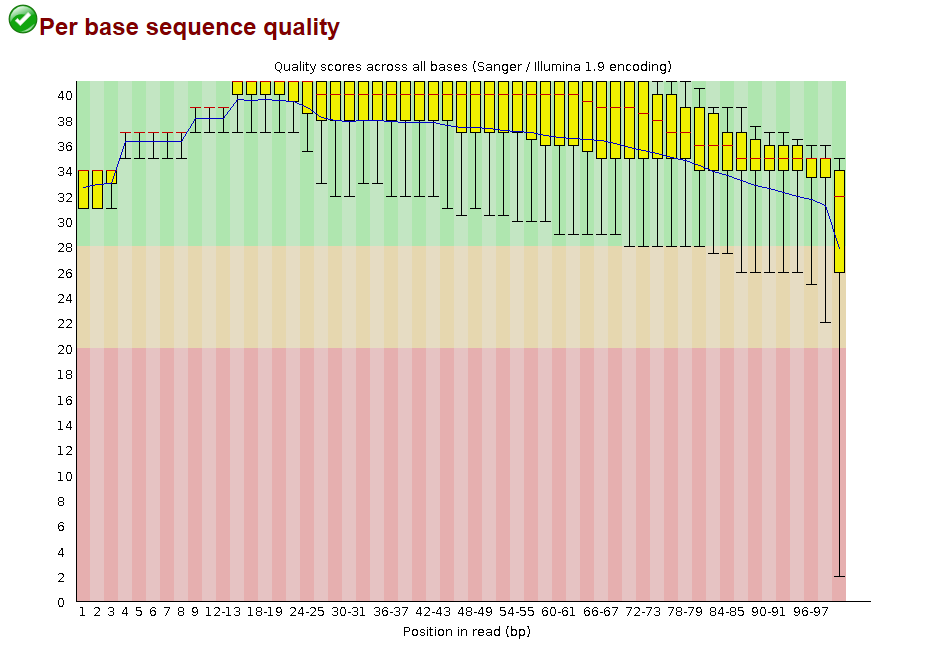 Качество ридов до чистки
Качество ридов до чистки
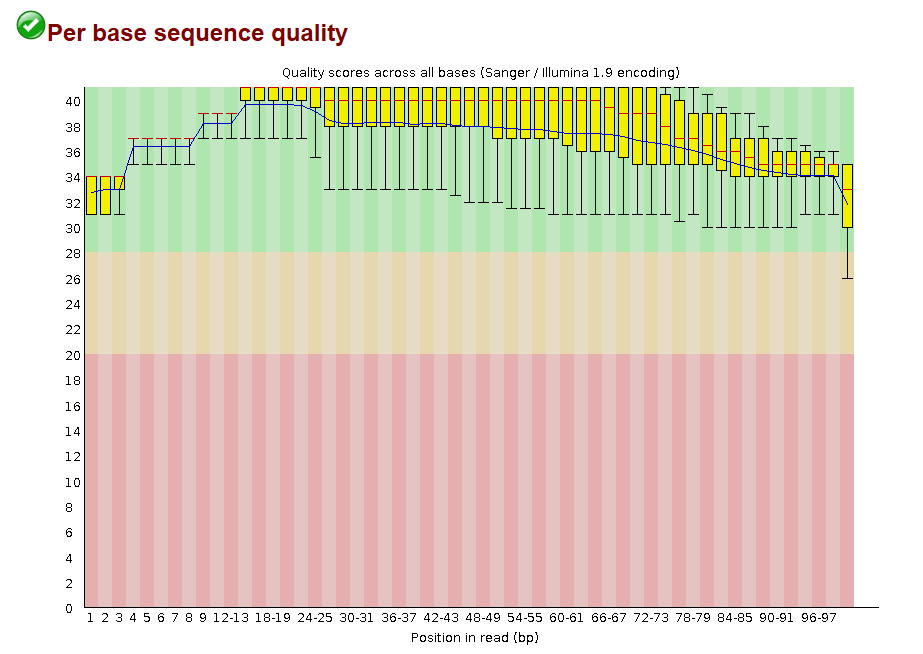 Качество ридов после чистки
Качество ридов после чистки
До чистки 10701, после 10630. Такое явление можно объяснить тем, что во время чистки все плохие риды полностью удаляются (тк в выдаче присутствуют такие, качество которых на протяжении всей длинны хуже 28). Концы контигов отрезаются тк "рамка" в 50 нуклеотидов движется вдоль рида, а потом рано или позно утыкается в участок который имеет плохое качество и затем программа отрезает все что идет после рамки.
Все операции были выполнены с помощью данных команд:
hisat2-build -f chr9.fasta chr9
индексируем референсную последовательность записанную в файл (chr9.fasta)
hisat2 --no-spliced-alignment --no-softclip -x ./ht2/chr9 -U chr9_1.fastq -S kart_normal.sam
картируем чтения и записываем выход в файл с форматом .sam (kart_normal.sam) (при этом картируем без разрыва и подрезания чтений)
число чтений картированных на хромосому 10553
число чтений не картированных на хромосому 148
Hisat2 выдает еще количество ридов, которые пытались наложить, количество ридов, которые наложились, которое не наложились, количество ридов, которые наложились больше одного раза и ровно 1 раз покрытие генома ридами.
Все операции были выполнены с помощью данных команд:
samtools view -b kart_normal.sam -o kart_normal.bam
переаводит формат sam в bam
samtools sort -f kart_normal.bam out.bam
сортируем по началу чтений, выходной файл bam (-f означает что задаем полностью имя выходного файла)
samtools index out.bam
индексируем отсортированный bam (на выйходе формат файла out.bam.bai)
1. кордината;4985879 тип полиморфизма: замена, что было в референсе, что найдено в чтениях;A ->G глубина покрытия данного места;30 качество чтений в данном месте.225.009
2. кордината;5122854 тип полиморфизма:, делеция что было в референсе, что найдено в чтениях;agg ->ag глубина покрытия данного места;DP=11 качество чтений в данном месте.95.4652
3. кордината;136132908 тип полиморфизма: вставка; что было в референсе, что найдено в чтениях;T ->TC глубина покрытия данного места; DP=29 качество чтений в данном месте.214.458
Все операции были выполнены с помощью данных команд:
samtools mpileup -uf chr9.fasta -g -o poli.bcf out.bam
теперь смотрим полиморфизмы. на входе файл с последовательностью хромосомы и bam файл, в котором все отсортированно по началу чтений
bcftools call -cv -o diff.vcf poli.bcf
показываем различия между полиморфизами в ридах и в реф последовательности
convert2annovar.pl -format vcf4 diff.vcf > diff.annovar
для аннотации необходима перевести формат в тот формат который читается программой
annotate_variation.pl -out refgene -build hg19 diff.annovar /nfs/srv/databases/annovar/humandb/
refgene - gene-based annotation: данная база позволяет посмотреть на возможные функции замен, на каких участках они происходят, изменения в аминокислотах
annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 diff.annovar /nfs/srv/databases/annovar/humandb.old/
dbsnp - filter-based annotation: отсюда получаем сколько snp имеют rs
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000gen diff.annovar /nfs/srv/databases/annovar/humandb.old/
1000 genomes - filter-based annotation
annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog diff.annovar /nfs/srv/databases/annovar/humandb.old/
Gwas - region-based annotation: клиническая аннотация
annotate_variation.pl diff.annovar /nfs/srv/databases/annovar/humandb.old/ -filter -dbtype clinvar_20150629 -buildver hg19 -out clinvar
Clinvar - filter-based annotation
Сводная google-таблица всех полиморфизмов (лист под названием "все")
Ответы на вопросы:
1.snp 102 инделей 5
2.Хорошее ли покрытие и качество у найденных полиморфизмов? Не у всех хорошее покрытие максимальное найденное 98 а самое маленькое 1 в основном покрытие маленькое, но есть и полиморфизмы с большим покрытием. ЖДумаю данным где есть большое покрытие можно доверять, и сказать что это действительно полиморфизм. но в основном там ошибка прочтения при этом качество тоже очень разное в основном плохое, но есть и достоверные полиморфизмы.
3.На какие категории делит snp база данных refseq в annovar? Сколько snp у Вас попало в каждую группу? refgene делит snp на несколько категорий: экзонные варианты в кодирующей последовательности snp(у меня их 17) интронные варианты которые попали в интроны snp (79) UTR3 варианты перекрывшиеся с 3’ некодируемом регионе (8) snp принадлежащие некодирующей области интергенетические (3)
4.В какие гены попали Ваши snp? JAK2 (Janus kinase 2) IL33 (interleukin 33) ABO (alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase)
5.К каким нуклеотидным и аминокислотным заменам привели snp?
есть замены в экзоме синонимичные (2) но у них нет rs, есть и несинонимичные(1) также у меня есть замены чья функция не установлена (14) (из выдачи refgene)6.Сколько snp имеет rs? у 95 есть rs (добываем из выдачи dbsnp)
7.Что Вы можете сказать о частоте найденных snp? Частота встречаемости snp примерно 1 к 357 нуклеотидам (Это значение медианы полученное так: сначала просчитано расстояние от каждой snp до следующей, а затем взята медиана всех значений.)
8.Что Вы можете сказать о клинической аннотации snp?