| команда | вход | выход | Что делает команда | Характеристики подготовки |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 SRR4240356.fastq outtrim.fastq ILLUMINACLIP:adapters.fa:2:7:7 | сырые чтния в файле SRR4240356.fastq, адаптеры сложенные в 1 файл adapters.fa | чтения с вырезанными адаптерами outtrim.fastq | удаляет адаптеры с ридов | было чтений 7511529 , стало чтений 7358424 (удалено 153105) |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 outtrim.fastq outtrim_short.fastq SLIDINGWINDOW:30:20 | outtrim.fastq чтения с вырезанными адаптерами | outtrim_short.fastq чтения очищенные от адаптеров и нуклеотидов низкого качества | сканирует чтения окном длинной 30 нукл, удаляет плохие буквы чье качество ниже 20 с концов чтений | удалено -238470 чтений; |
| в общем удалено: 391575 | ||||
| размер файла до чистки 793999866 | ||||
| размер файла после чистки: 749258208 | ||||
| размер отличается на 44741658 байт |
| velveth velveth.dir 29 -fastq outtrim_short.fastq -short | velveth.dir директория куда направляется весь выход | очищенные риды outtrim_short.fastq | располагает риды по порядку | "получилось три файла Log Roadmaps Sequences |
| в log прописывается версия." |
| velvetg velveth.dir | velveth.dir директория с выходом команды, которая нарезает к-меры | ||
| N50 | 73133 | ||
| 3 самых длиных контига | |||
| 7 | 115468 | 52.217359 | |
| 19 | 106076 | 45.970578 | |
| 8 | 75082 | 54.507059 | |
| описание аномально больших или малых покрытий | |||
| типичное покрытие | 634,8946654 | ||
| 1 | покрытие | длина | id |
| 358 300,00 | 1 | 497 | |
| 2 | 1,74 | 19 | 121 |
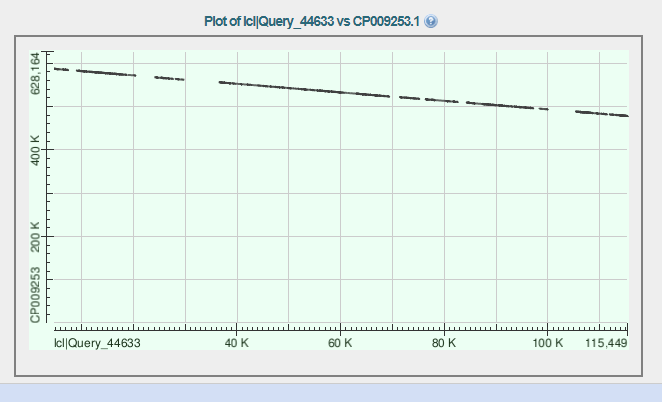
Dot Matrix View самого длинного контига
| 1. | нач | кон | |
| координаты | 478095 | 584329 | |
| хар-ка выравниваний (было 17 шт)данные приведены для 1 | гэп | число различий | |
| 545 | 4026 | ||
| Хар-ка полного выравнивания | Покрытие контига | процент индентичности | вычисленное число различий |
| 73 | 81 | 16015 пар оснований | |
| как ложиться | число совпадений | на картинке видно что ложится ровно. Есть выпавшие участки. самый большой выпавший участок это примерно 500 нуклеотидов | |
| 17 | количество больших выпавших кусков 8 |
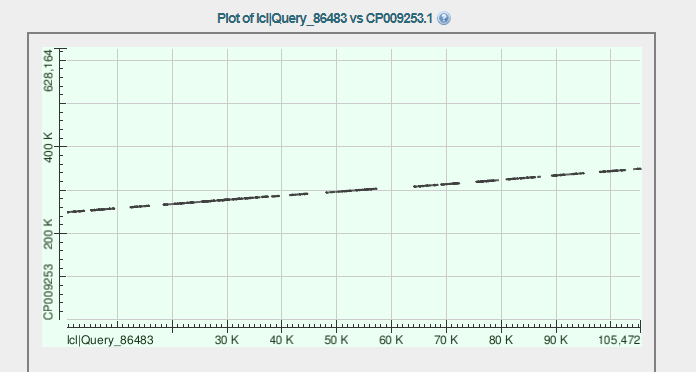
Dot Matrix View второго по длине контига
| 2. | нач | кон | |
| координаты | 248967 | 349674 | |
| хар-ка выравниваний (20 шт) данные приведены для первого | гэп | число различий | |
| 94 | 719 | ||
| Хар-ка полного выравнивания | Покрытие контига | процент индентичности | вычисленное число различий |
| 68 | 79 | 15148 пар оснований | |
| как ложиться | число совпавших участков | на картинке видно что есть выпавшие участки, максимальный выпавший участок это 700 пар нуклеотидов | |
| 20 | общее количество внушительных выпавших кусков 11 |
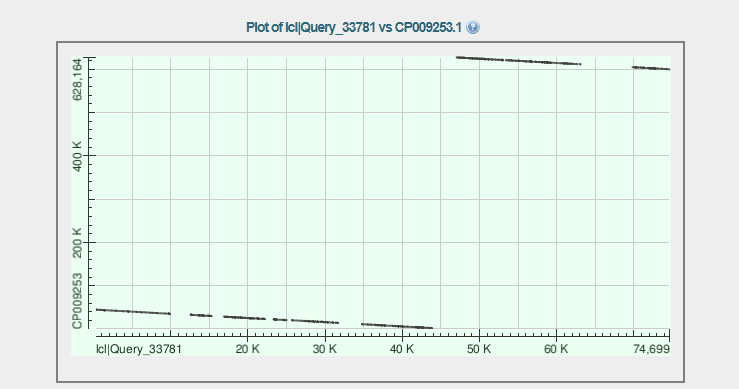
Dot Matrix View третьего по длине контига
| 3. | нач | кон | |
| координаты | 2004 | 621055 | |
| хар-ка выравнивания одного | гэп | число различий | |
| 252 | 1992 | ||
| харка полного выравнивания | Покрытие контига | процент индентичности | вычисленное число различий |
| 73 | 83 | 9317 | |
| как ложиться | число совпавших участков | на картинке видно что контиг поделился на две примерно одинаковые части, а значит алгоритм сборки de nova дал сбой и соединил не нужные учистки. | |
| 13 | ну или же организм откуда проба-мутант (но это маловероятно) | ||
| количество больших выпавших участков: 7 | |||
| максимальная длинна выпавшего участка 300-400 нуклеотидов (смотрим по нижнему куску выровненного гэпа) |