1.Таксономия и функция прочтённой нуклеотидной последовательности (из практикума 6)
используйте BLASTN ("Somewhat similar sequences") по банку nr
(i) предполагаемую функцию
cytochrome oxidase subunit 1 (COI) gene, partial cds; mitochondrial. (Ген 1 субъединицы оксидазы цитохрома)
Полученная последовательность несет данную функцию потому что во всей выдачи
программы бласт все гомологичные последовательности несут такую функцию
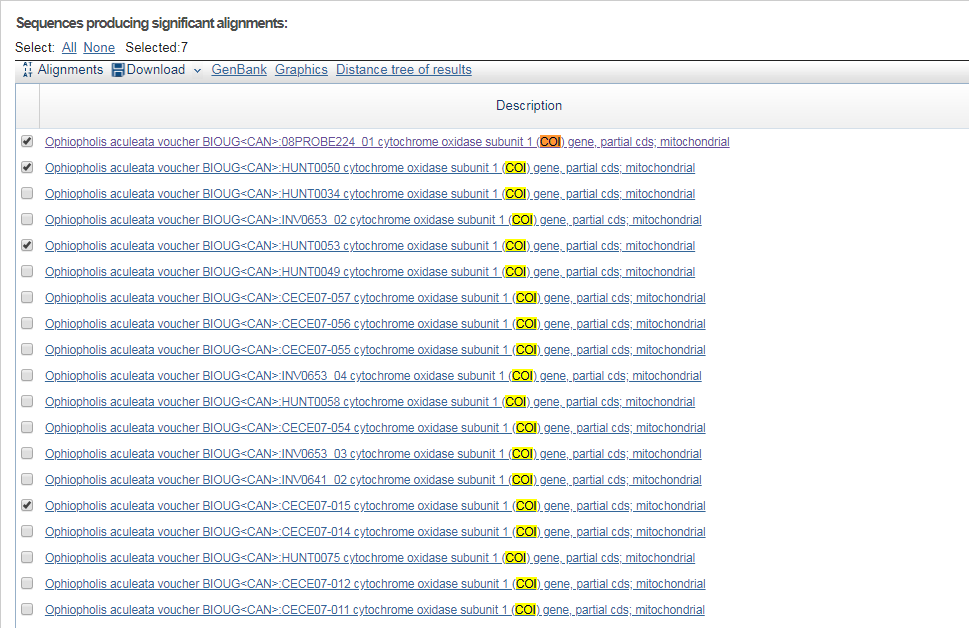 (фото почемуCOI)
(фото почемуCOI)
1.PNG
(ii) предполагаемую таксономию;
передо мной явно стояла легче задача, тк в выдаче программы (те первых 100 наиболее достоверных выравниваний ) найдены в одном и том же генусе
Ophiopholis.
Ophiopholis aculeata (в основном среди наиболее достоверных) диапазон весов:1100- 976
japonica 721
kennerlyi 654-676
из 10 возможных известных в blast (Ophiopholis aculeata (daisy brittle star)
Ophiopholis japonica
Ophiopholis kennerlyi
Ophiopholis longispina
Ophiopholis sp. EAC01
Ophiopholis sp. GP0048
Ophiopholis sp. GP0049
Ophiopholis sp. GP0050
Ophiopholis sp. GP0051
Ophiopholis sp. KP-2011
Уже по приведенным данным можно понять, чо выделенная последовательность скорее всего из вида Ophiopholis aculeata,
тк наиболее высокие веса выравниваний соответствуют пробам именно из этого вида, но для достоверности выровняем последовательности внутри этого генуса,
чтобы посмотреть уровень замен в этом генусе в разных видах.
я построила прект в jalview выровненных гомологичных последовательностей.
таксон.jvp
отсюда видно что сгенерированная нами последовательность (my_seq) вероятнее всего была выделена из O.aculeata.
Какие находки достаточны для аннотации:
(они немного различаются потому что взяты из одного организма но разными пробами)
для примера возьмем также одну изолированнную пробу (MT07097)
для разнообразия возьмем еще находку с процентом инд 86-котоорая просто относится к другому виду HM473933.1
Вспомогательные материалы:
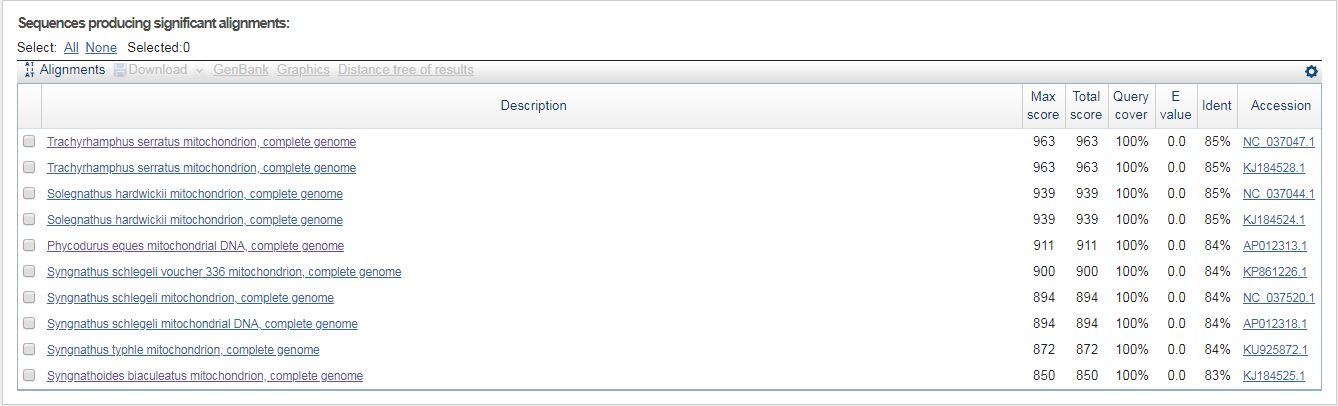
Таблица1:это "опорные выдачи программы бласт для аннотации данной последовательности.
Основное в этой таблице то, что вся выдача разная, но несет одинаковую функцию.
|
Max score |
Total score |
Query cover |
E value |
Ident |
Accession |
комментарии |
| 1 |
1100 |
1100 |
100% |
0.0 |
99% |
HM473856.1 |
первое-наилучшее |
| 2 |
1095 |
1095 |
100% |
0.0 |
99% |
HM542291.1 |
с макс скор чуть меньше |
| 3 |
1086 |
1086 |
100% |
0.0 |
99% |
HM542294.1 |
следующая ступень макс скора |
| 4 |
1081 |
1081 |
99% |
0.0 |
99% |
HM542285.1 |
первое падение покрытия |
| 5 |
1068 |
1068 |
100% |
0.0 |
98% |
HM473852.1 |
первое падение процента индентичности до 98 |
| 6 |
1028 |
1028 |
100% |
0.0 |
97% |
KJ620598.1 |
изолированная проба |
| 7 |
721 |
721 |
100% |
0.0 |
86% |
HM473933.1 |
наилучшее выравнивание с другим видом (тем же родом) |
Таблица2: Необходимая выдача, которая понадобилась для определения вида.
|
Description |
Max score |
Total score |
Query cover |
E value |
Ident |
Accession |
| 1 |
Ophiopholis aculeata voucher BIOUG:08PROBE224_01 cytochrome oxidase subunit 1 (COI) gene, partial cds; mitochondrial |
1100 |
1100 |
100% |
0.0 |
99% |
HM473856.1 |
| 2 |
Ophiopholis japonica voucher BIOUG:HLC-24059 cytochrome oxidase subunit 1 (COI) gene, partial cds; mitochondrial |
721 |
721 |
100% |
0.0 |
86% |
HM473933.1 |
| 3 |
Ophiopholis kennerlyi voucher 10BIOBC-EC023 cytochrome oxidase subunit 1 (COI) gene, partial cds; mitochondrial |
676 |
676 |
100% |
0.0 |
84% |
KU495789.1 |
2. Сравнение списка находок нуклеотидных последовательностей тремя разными вариантами blast
Возьмите сначала ту же последовательность, что в задании 1,
В отчете укажите все параметры каждого запуска BLAST.
Параметры запуска бласт:
| Optimize for |
Highly similar sequences (megablast) |
Somewhat similar sequences (blastn) |
Somewhat similar sequences (blastn)(sence) |
| Database |
nr |
nr |
nr |
| Word size |
28 |
11 |
7 |
| Max target sequences |
10 |
10 |
10 |
| Max matches in a query range |
1 (1 выдача ), 3 (6 выдач), 0 |
0 |
0 |
| Match/Mismatch Scores |
1,-2 |
2,-3 |
2,-3 |
| строчка с исключением рода |
Ophiopholis (taxid:35051) |
Exclude |
|
|
Gnathophiurina (taxid:41168) |
|
|
| Число находок: |
152 |
2159 |
2292 |
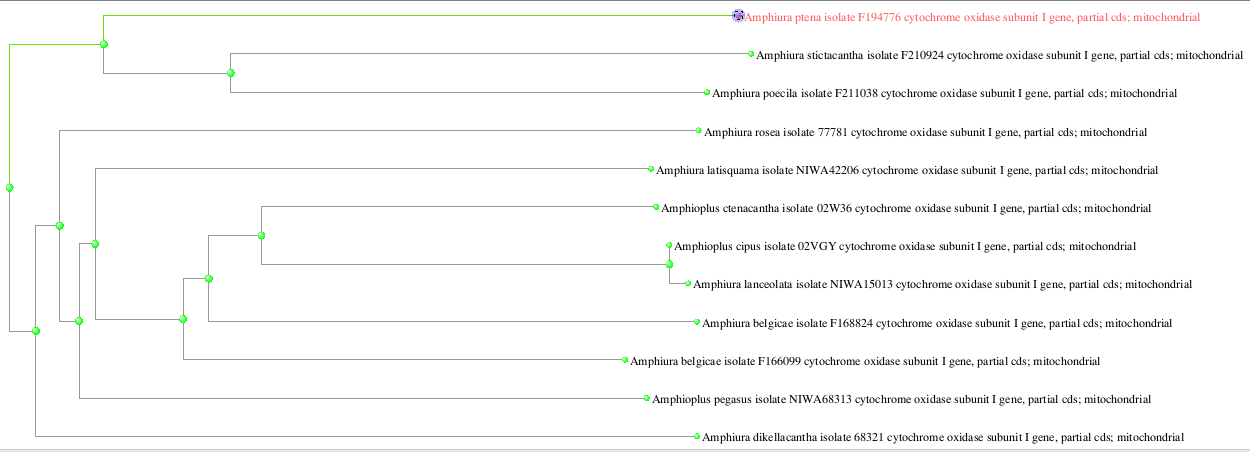
для наглядности я сравнивала деревья (_tree)
как и следовало ожидать в чувствительном и не очень результаты индентичны
 2.PNG чувствит
2.PNG чувствит
 3.PNG не очень
3.PNG не очень
 4.PNG а megablast отличился.
4.PNG а megablast отличился.
картинки:
EMBOSS_001-наша последовательность. комментарий к картинкам
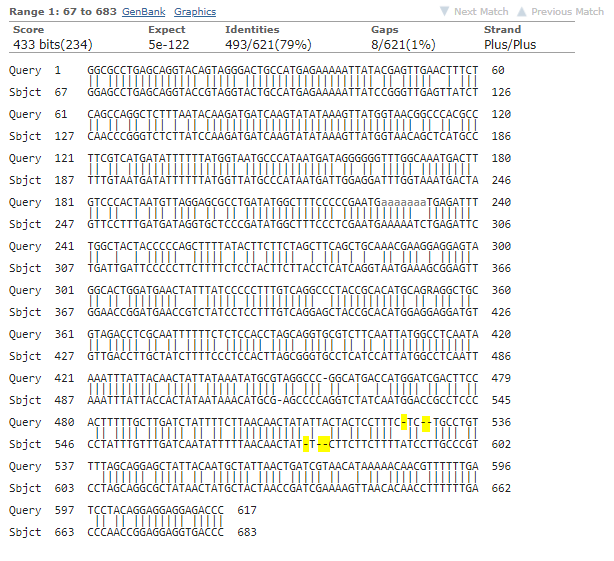
сравним какую нибудь находку присутствующую во всех трех выдачах (10 ограничение) (розовая рамочка картинки 3 и 4)
например:
KU895415.1
в чувствительной e-val чуть выше чем в обычной
а вот в megablast выравнивание отличается на один гэп.
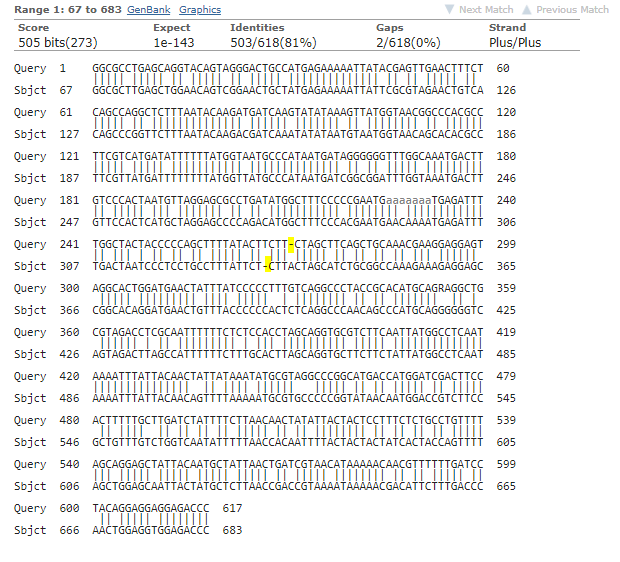 5.PNG выравнивание отлич на 1 гэп из мегабласта
5.PNG выравнивание отлич на 1 гэп из мегабласта
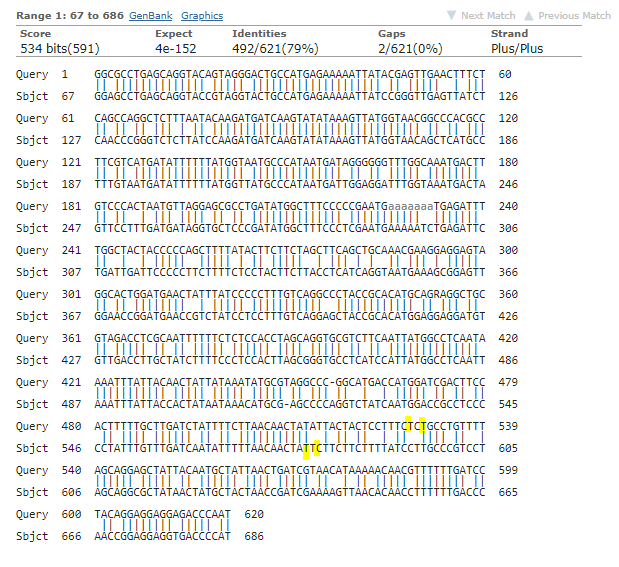
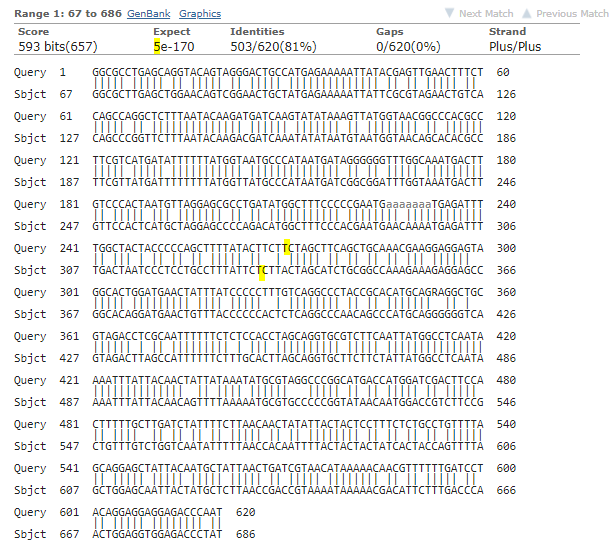 6.PNG выравнивание чувствительным методом и помечено отличие в и валью
6.PNG выравнивание чувствительным методом и помечено отличие в и валью
Думаю что разница в выравниваниях объясняется разными целями алгоритмов.
Blastn как было сказано выше ищет схожие последовательности, те если последовательность
гомологична то один гэп погоды не сделает. а вот если алгоритм ищет место нахождения в геноме (megablast) то один гэп меняет место нахождения.
поэтому в заданных параметрах в бластн не может быть линейного штафа за гэп, как в мегабласте.
Сравнение деревьев заставило меня обратить внимание на одно из выравниваний алгоритма
megablast - потому что это выравнивание не нашел blastn на ограничении 10(хотя должен находить дальних родственников)
KU895034.1
ссылка на голубую рамочку 4.PNG в мегабласте.
если расширить поиск во всех остальных алгоритмах ДО 250(!!) то они его предлагают. Это очень
странно, ведь мегабласт вроде указал что последовательность очень похожа, но при этом бласт н
отказался включать ее в “родственников” и предлагать данный вид как вариант откуда могли взять последовательность.
#переделай деревья#
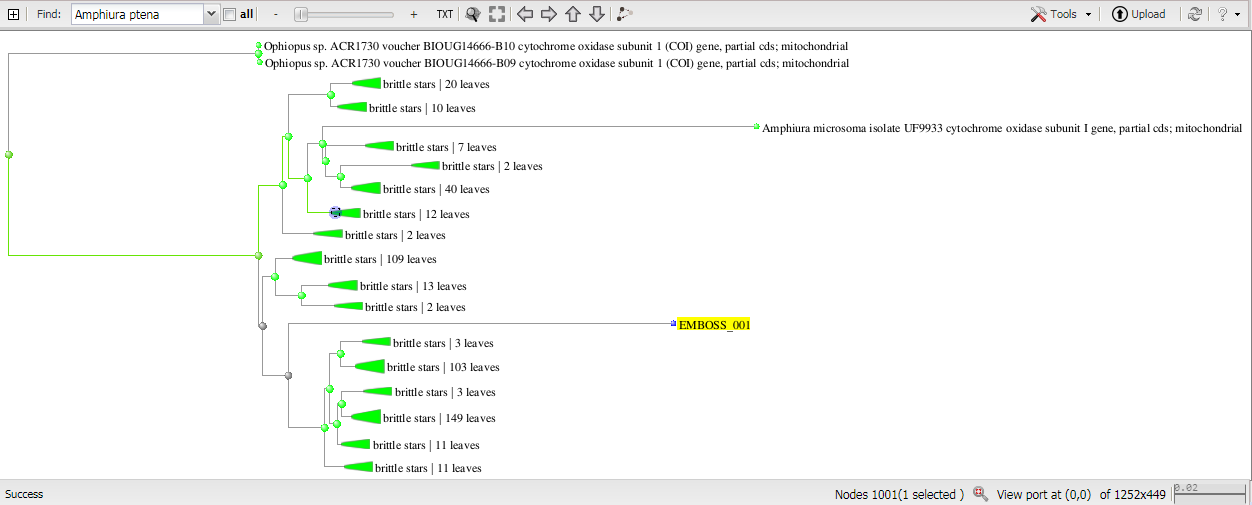 7.PNG полное
7.PNG полное
7.PNG полное
 8.PNG ветвь
8.PNG ветвь
Расширенное до 500 дерево и нахождение внутри него находки мегабласта
Если сравнить выравнивания то опять натыкаемся н то что мегабласт приветствует гэпы а бластн нет, возможно по вышесказанной причене
Комментарий по гэпам:
 9.PNG присутствие гэпа в мегабласт
9.PNG присутствие гэпа в мегабласт
 10.PNG отсутствие гэпа в бластн
10.PNG отсутствие гэпа в бластн
Из такого большого разнообразия организмов предлагаемых бластн, перед тем как выдать
схожую (по мнению мегабласта ) последовательность можно заключить что параметры, по которым действует
алгоритм бластн действительно помогают отобрать организм, из которого выделена проба. По сравнению с мегабластом, который ищет место в геноме.
Митохондриальное РНК
| Optimize for |
Highly similar sequences (megablast) |
Somewhat similar sequences (blastn) |
Somewhat similar sequences (blastn)(sence) |
| Database |
nr |
nr |
nr |
| Word size |
28 |
11 |
7 |
| Max target sequences |
10 |
10 |
10 |
| Max matches in a query range |
1 (1 выдача, ) 3 (6 выдач) 0 |
0 |
0 |
| Match/Mismatch Scores |
1,-2 |
2,-3 |
1,-4 |
| строчка с исключением рода |
Hippocampus (taxid:72046) |
Exclude |
|
|
Syngnathoidei (taxid:1489884) |
|
|
| Число находок: |
191 |
312 |
391 |
все выдачи у чувствительного и мега стыкуются кроме последнего.KJ184525.1 мегабласт и чувствительный KJ139455.1 бластн
картинки выдачи:
 11.PNG мегабласт
11.PNG мегабласт
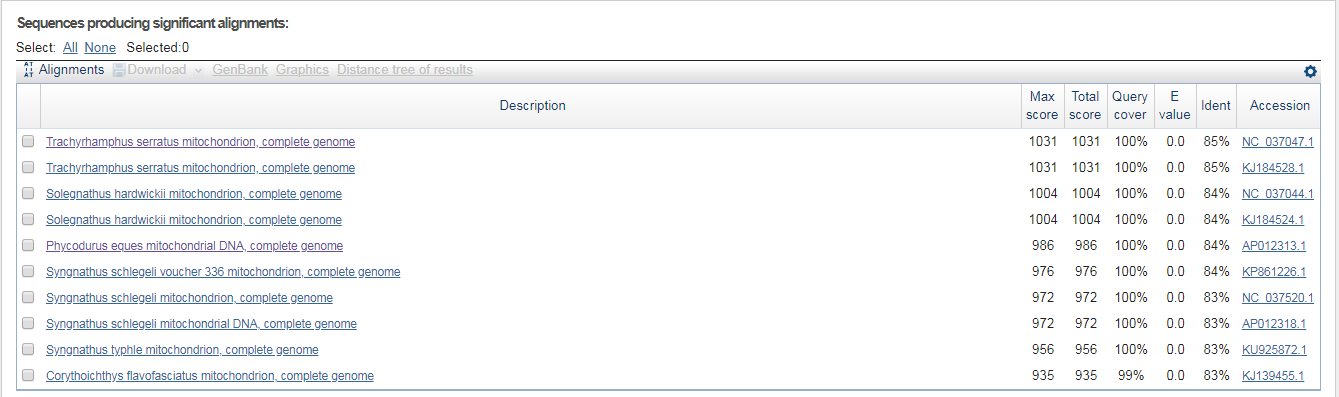 12.PNG бластн
12.PNG бластн
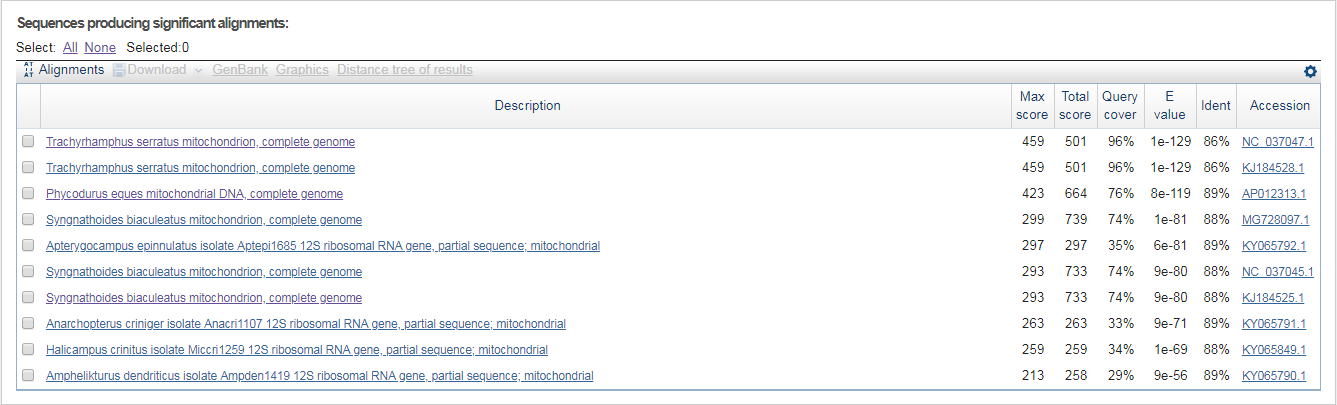 13.PNG бластн чувствительный
13.PNG бластн чувствительный
в Выдаче программы если обзорно посмотреть на выравнивания, в них прослеживаются те же тенденции поведения алгоритмов.
но как я должна заметить в митохондриальных геномах все алгоритмы дают очень похожую выдачу.
Из этого следуют такие выводы:
Чем более схожую выдачу дают разные алгоритмы поиска выравниваний, тем возможно более консервативными являются гены.
По сути дела разные алгоритмы направлены на поиски одного и того же - выравниваний с гомологичными последовательностями (даже когда ищем
место в геноме-выравниваем предположительно одни и те же последовательности, но чуть измененные). Получается чем меньше видоизменен
ген тем более вероятно мегабласт найдет его в родственном
организме. Здесь и происходит состыковка двух алгоритмов (нахождения родственников-поиск места в геноме)
почти одинаковая выдача программ мегабласта и бластн говорит о том что митохондриальный геном более консервативен чем предыдущий ген.
Оно и понятно-это же рибосомальное РНК, где почти все нуклеотиды повторяются у разных организмов, что уже указывает на консервативный остов.
Но если посмотреть на деревья, то одни и те же последовательности разные алгоритмы
выстраивают по разному. конечные ветви у этих деревьев схожи, но между ними устанавливаются родственные отношения по разному.
Любопытно что мегабласт и бластн чувствительный вставляют исходную последовательность в разные места дерева
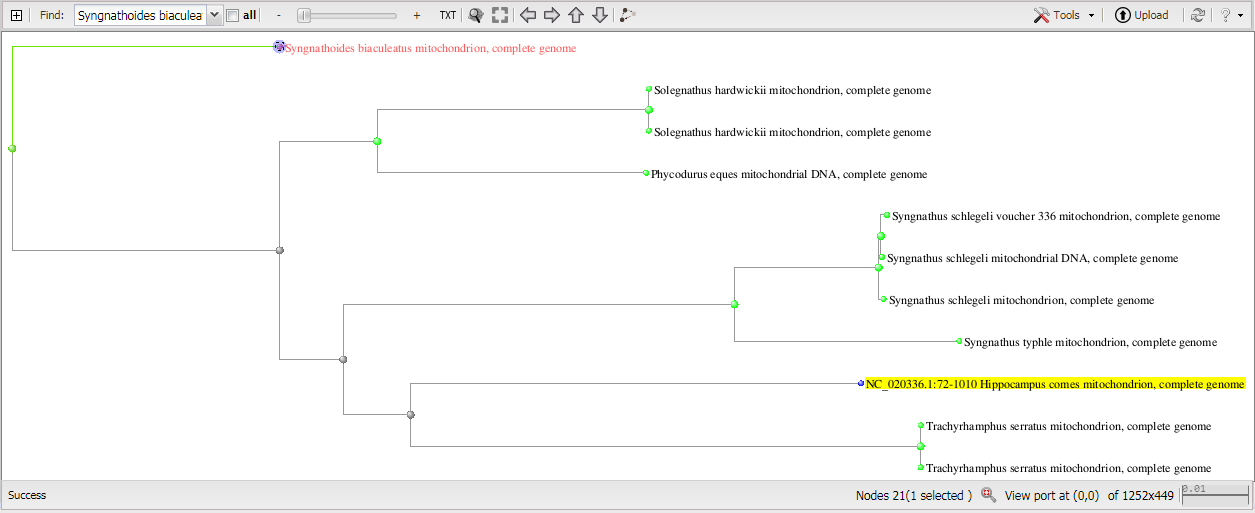 14.PNG мегабласт
14.PNG мегабласт
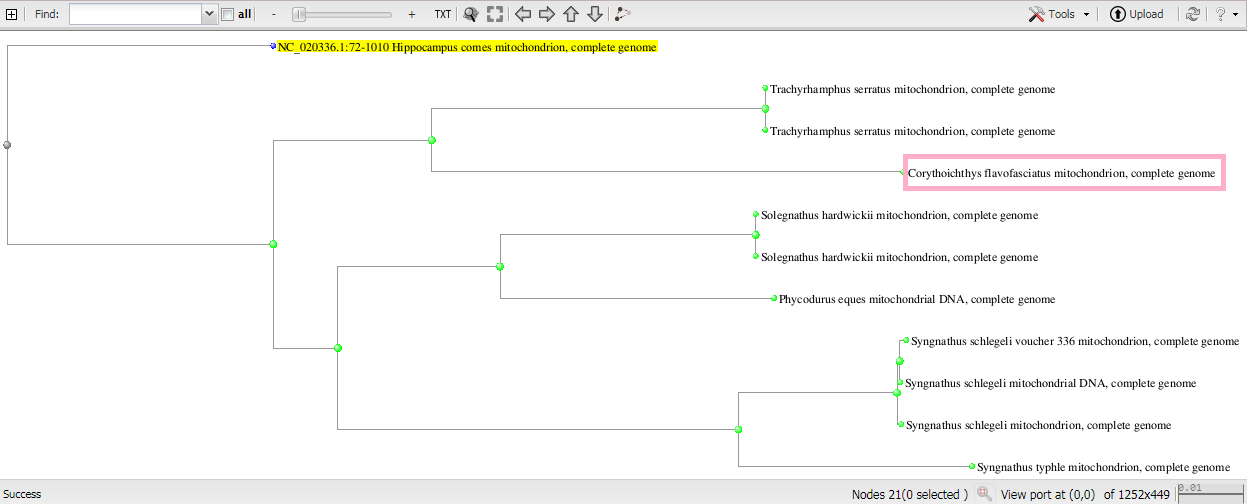 15.PNG бластн
15.PNG бластн
Розовым цветом помечено единственное различие в выдачи.
Если рассмотреть выдачу чувствительной программы бластн
она содержит первые две выдачи, акие же что и все остальные алгоритмы, затем на третьем месте у нее выдача которая в бластн и мегабласт стоит 5
Еще одна из выдач алгоритма бластн чувствительного совпадает с выдачей мегабласта но не совпадает с выдачей бластн.
те у чувствительного метода 4 выдачи совпадают с мегабластом и 3 выдачи совпадают с бластн.
если посмотреть на выравнивания одной и той же последовательности KJ184525.1 но предлагаемые разными алгоритмамми (бластн чувствительный и мегабласт)
 16.PNG мегабласт
16.PNG мегабласт
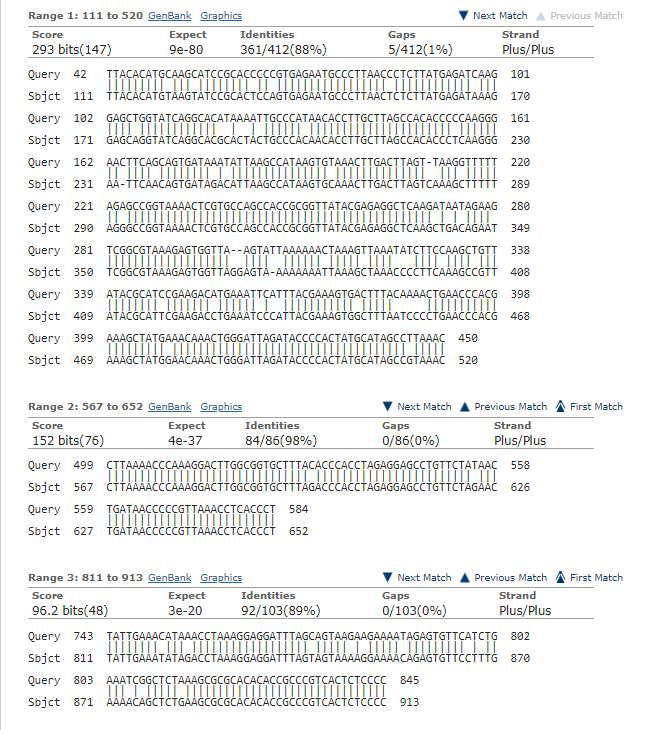 17.PNG чувств1
17.PNG чувств1
 18.PNG чувств2
18.PNG чувств2
Алгоритм бластн позволяет себе дробить выравнивание. Думаю связано это опять же с тем что на выдаче алгоритма подразумеваются гомологичные последовательности. То
есть с ходом эволюции в последовательности могут возникать вставки. И чтобы программа выдавала гомологию на выходе не смотря на какие то преобразования в ходе эволюции
алгоритм "умеет" дробить последовательность.этого не дозволено мегабласту, тк по факту мы должны знать уже организм чей ген мы пытаемся выровнять, и нам нужно найти
место, а значит никаких существенных изменений в последовательности быть не должно. (Помимо ошибок прочтения)
3.Проверка наличия гомологов трех белков в неаннотированном геноме
длина скаффолдов в геноме = 23,962,143
На мой взгляд таблица в формате по умолчанию удобнее тк выдает все выровненные скэффолды, а в таблицу выдает по порядку размещенные данные
и убирает неинформативные данные. Если задать к качествфе выводного формата таблицу, то в таблицу будут включены лучшие выравнивания+ выравнивания отдкльных
кусочков каждого скэффолда не смотря на вес.
1.Найдено 22 выравнивания среди неаннотированного генома
для HSP71_YEAST P10591 Heat shock protein SSA1 (Heat shock protein YG100) белка теплового шока у дрожжей.
пару фразо белке
длинна = 642;
Среди находок явно есть гомологи, где e-value =0.0 (возможно такая "точность" возникает изза очень маленькой длинны базы данных)
query id subject id % identity, alignment length, mismatches, gap opens, q. start, q. end, s. start, s. end, evalue, bit score
(1.1)HSP71_YEAST scaffold-199 78.98 609 125 2 2 607 1109256 1107430 0.0 920
HSP71_YEAST scaffold-96 66.06 607 201 4 3 606 89928 91742 0.0 744
HSP71_YEAST scaffold-423 65.73 607 203 4 3 606 1313216 1311402 0.0 737
(1.2)HSP71_YEAST scaffold-199 55.56 27 12 0 82 108 1110027 1109947 0.002 40.8
Видно, что белок выровнялся почти по всей длинне 94,5% в находке1.1. К тому же, в первой находке есть еще и маленький домен длинной 27 нуклеотидов (1.2).
Получается, что невыровненной длинны всего 0.93% от общей длинны. Учитывая вес выравнивания (920) и evalue, стемящееся к 0, можно почти наверняка сказать,
что белок сохраняет свои функции и в Amoeboaphelidium protococcarum.
2. TERT_SCHPO O13339 Telomerase reverse transcriptase
теломераза,восстанавливающая длину хромосомы при репликации; имеется у большинства (но не всех) эукариот;
длина белка =988
найдено 5 выравниваний
Среди них есть условные гомологи:
query id subject id % identity, alignment length, mismatches, gap opens, q. start, q. end, s. start, s. end, evalue, bit score
TERT_SCHPO unplaced-307 26.84 503 282 17 320 780 14863 16239 7e-22 102
TERT_SCHPO scaffold-17 25.05 491 305 16 320 780 610900 612273 1e-23 108
Их можно считать условными гомологами тк длина выровненной последовательности это 503 491 (те 51% и 50% от всей длинны белка), и к тому же значения evalue
тоже маленькие, значит какие-то отрывки этого белка все же экспрессируются.
Но с другой стороны тут настораживает значения процента индентичности который во всех длинных выравниваниях составляет порядка 1/4. Процент индентичности двух
случайно перемешанных последовательностей тоже составляет 1/4. Значит выравнивание программой могло быть ошибочным и не указывающим на гомологию.
Одно можно сказать наверняка: в геноме присутствует один домен этого белка длинной 36 аминокислот. У этого домена процент индентичности 42%, но тут вылезает
очень большой e-value. Как я понимаю, когда мы берем как базу данных один геном, e-value значение может быть не показательным, в силу маленькой общей длинны db..
TERT_SCHPO unplaced-647 41.67 36 21 0 170 205 141 248 4.9 28.1
3.MCES_ENCCU Q8SR66 mRNA cap guanine-N7 methyltransferase
белок метилтрансфераза гуанина N-7 КЭП матричной РНК выделенный из Encephalitozoon организма из того же царства (Fungi) что и Amoeboaphelidium protococcarum.
Длина =283
Программа нашла 2 выравнивания, одно из которых:
MCES_ENCCU scaffold-550 28.81 243 142 5 5 218 47988 48710 3e-12 66.2
Одно из находок покрыло 243 аминокислоты (86%) длины и имеет e-value 3e-12, но тк все ранво процент индентичности = 28, его можно считать условно гомологом.
4.Ген белка в одном из контигов в неаннотированном геноме
scaffold-158 нашлись очень маленькие участки перекрывания
scaffold-67
nplaced-1011 (9672 letters)
Рассмотренные белки:
| Name |
Max score |
Total score |
Query cover |
E value |
Ident |
Accession |
| amino acid permease [Coprinopsis cinerea okayama7#130] |
89.4 |
89.4 |
10% |
2,00E-15 |
30% |
XP_001832190.2 |
| hypothetical protein SPPG_05591 из Spizellomyces punctatus DAOM BR117 |
143 |
143 |
14% |
2,00E-32 |
33% |
XP_016607384.1 |
Нашлись известные белки, такие как:
amino acid permease [Coprinopsis cinerea okayama7#130]
процент индентичности выше чем в случайных выровненных последовательностях
e-value порядка 10 в минус 15 степени
белок имеет длинну 740 аминокислот, при этом выровнялись 316 (43%) при выравнивании возникли большие гэпы, что может быть связано с интронами.
Сложно сказать, что это ген белка пермиазы аминокислоты.
:изображение выравнивания:
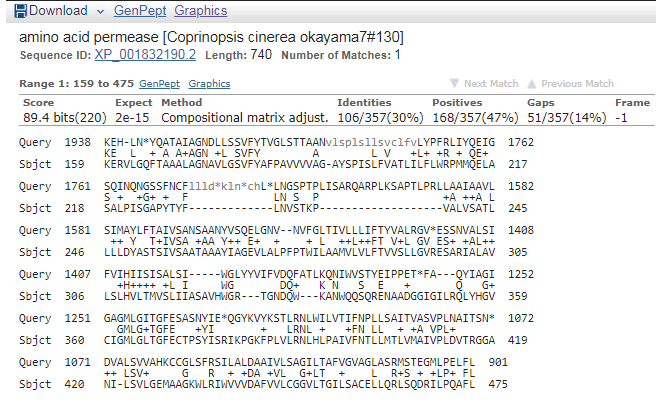 19.PNG
19.PNG
Кроме того среди выдачи нашлись и гипотетические белки с лучшими выравниваниями, например
hypothetical protein SPPG_05591 из Spizellomyces punctatus DAOM BR117
143: 143: 14%: 2e-32: 33%: XP_016607384.1
Общая длинна белка 688 аминокислот, из которых 418 выравниваются с нашей последовательностью (61%)
при выравнивании аналогично возникает большой гэп, что так же вероятно связано с присутствием интронов
;картина выравнивания;
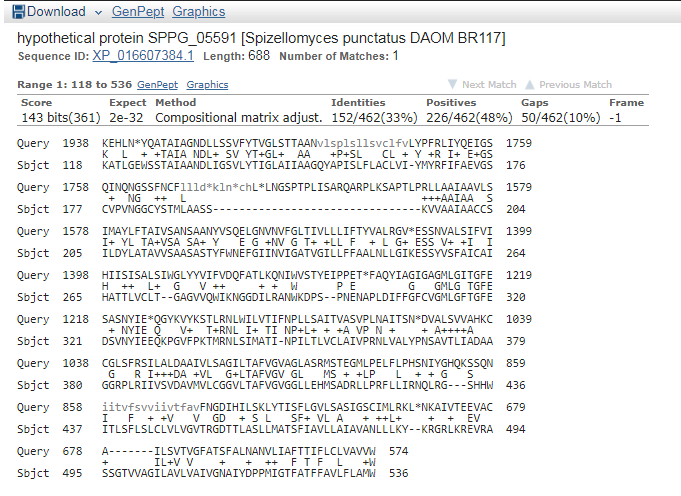 20.PNG
20.PNG
5.Карта локального сходства геномов двух бактерий
Неудачная попытка:
Chlamydia trachomatis 434/Bu chromosome, complete genome
NCBI Reference Sequence: NC_010287.1
Chlamydia gallinacea 08-1274/3, complete genome
NCBI Reference Sequence: NZ_CP015840.1
бедная картинка
Удачная попытка:
Chlamydia trachomatis D/UW-3/CX chromosome, complete genome
NCBI Reference Sequence: NC_000117.1
Chlamydia gallinacea 08-1274/3, complete genome
NCBI Reference Sequence: NZ_CP015840.1
Я получила сначала точечное представление по Megablast, но она показалась мне разрывной:
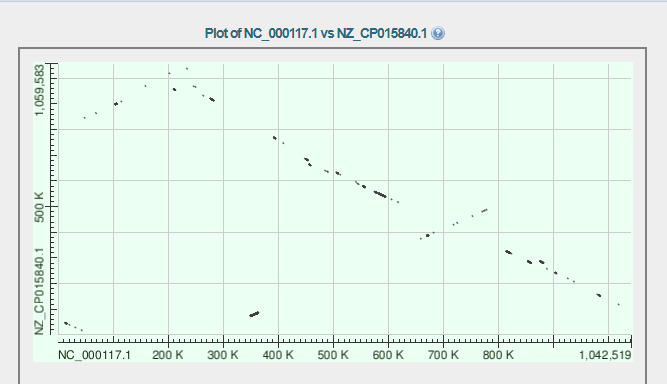 Megablast
Megablast
А потом я выровняла по механизму BlastN, картина оказалась гораздо более приятная, поэтому я все преобразования буду рассмотривать на ней
картина1_blastn
на выравнивании видно что два вида принадлежат одному роду, тк на протяжении всей длинны почти всегда стабильная линия выравнивания
на представлении точечной матрицы вида одна инверсия
координаты
NC_000117.1 630К-800К соответствует NZ_CP015840.1 350К-500К
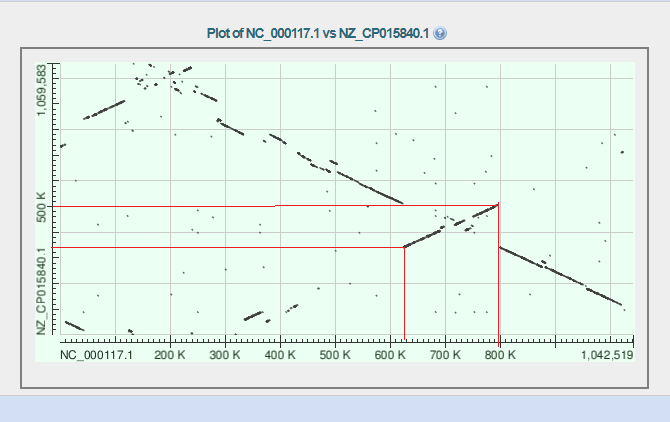 Инверсия
Инверсия
Здесь так же имеет место такая перестройка, как делеция
первая делеция участка происходит в NZ_CP015840.1 (в районе 780К) -участок который пропал соответствует координатам 330К-380К NC_000117.1.
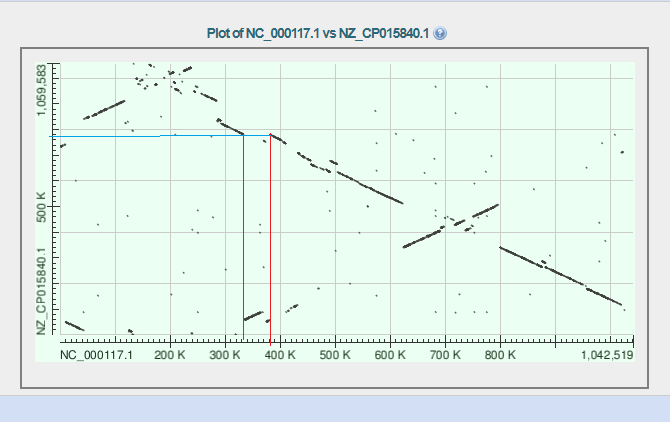 делеция
делеция
На карте так же заметна возможная транслокация
в NC_000117.1 ген который должен находиться в районе 270К осуществяет инверсию и переходит на участок 50К-120К
а в NZ_CP015840.1 этот ген соответствует участку 850К-900К
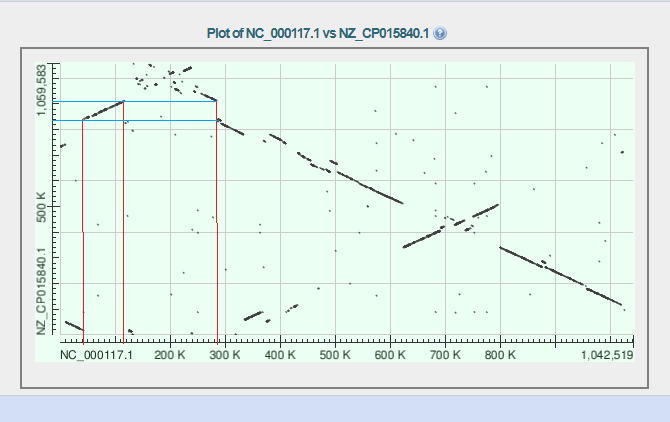 транслокация
транслокация
Учебная почта
© Бердникович Екатерина, 2017