Докинг низкомолекулярных лигандов в структуру белка
Цель данного занятия ознакомится с возможностями докинга низкомолекулярного лиганда в структуру белка.
Объект - белок лизоцим, структуру которого постронеа на основе гомологичного моделированияIn [4]:
#Модели
import numpy as np
import copy
# Отображение структур
import IPython.display
import ipywidgets
from IPython.display import display,display_svg,SVG,Image
# Open Drug Discovery Toolkit
import oddt
import oddt.docking
import oddt.interactions
# Органика
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
import pmx # Модуль для манипулирования pdb
Подготовка белка
In [6]:
pdb=pmx.Model('WithLigand.B99990001.pdb')
for r in pdb.residues[135:]:
print r
In [7]:
# создание объектов белок и лиганда
newpdb = pdb.copy()
lig = pdb.copy()
del newpdb.residues[226:]
del lig.residues[:226]
In [8]:
# был найден геометрический центр лиганда
x_c = 0
y_c = 0
z_c = 0
for a in lig.atoms:
x_c = x_c+a.x[0]
y_c = y_c+a.x[1]
z_c = z_c+a.x[2]
x_c = x_c / len(lig.atoms)
y_c = y_c / len(lig.atoms)
z_c = z_c / len(lig.atoms)
print "center ["+str(x_c)+", "+str(x_c)+", "+str(x_c)+"]"
In [9]:
newpdb.writePDB("ProteinDelResid.pdb")
Подготовка белка для докинга
In [10]:
prot = oddt.toolkit.readfile('pdb','ProteinDelResid.pdb').next()
prot.OBMol.AddPolarHydrogens()
prot.OBMol.AutomaticPartialCharge()
print 'is it the first mol in 1lmp is protein?',prot.protein,':) and MW of this mol is:', prot.molwt
Создание лигандов, где метильный радикал этой группы будет заменён на [OH,NH3+,H,Ph,COO-]
In [12]:
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit import RDConfig
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
import numpy as np
from IPython.display import display,Image
ibu=Chem.MolFromSmiles('CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O')
AllChem.Compute2DCoords(ibu)
display(ibu)
ibu1=Chem.MolFromSmiles('OC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O')
AllChem.Compute2DCoords(ibu1)
display(ibu1)
ibu1=Chem.MolFromSmiles('C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O')
AllChem.Compute2DCoords(ibu1)
display(ibu1)
ibu1=Chem.MolFromSmiles('[NH3+]C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O')
AllChem.Compute2DCoords(ibu1)
display(ibu1)
ibu1=Chem.MolFromSmiles('[O-]C(=O)C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O')
AllChem.Compute2DCoords(ibu1)
display(ibu1)
ibu1=Chem.MolFromSmiles('c2ccccc2C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O')
AllChem.Compute2DCoords(ibu1)
display(ibu1)






In [14]:
smiles = ['CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O', 'OC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O','C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O','[NH3+]C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O','[O-]C(=O)C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O','c2ccccc2C(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O']
mols= []
images =[]
for s in smiles:
m = oddt.toolkit.readstring('smi', s)
if not m.OBMol.Has3D():
m.make3D(forcefield='mmff94', steps=150)
m.removeh()
m.OBMol.AddPolarHydrogens()
mols.append(m)
###with print m.OBMol.Has3D() was found that:
### deep copy needed to keep 3D , write svg make mols flat
images.append((SVG(copy.deepcopy(m).write('svg'))))
display_svg(*images)
In [17]:
#create docking object
dock_obj= oddt.docking.AutodockVina.autodock_vina(
protein=prot,size=(20,20,20),center=[25.3029178982, 25.3029178982, 25.3029178982],
executable='/usr/bin/vina',autocleanup=True, num_modes=20)
print dock_obj.tmp_dir
print " ".join(dock_obj.params) # Опишите выдачу
Докинг и его результаты
In [18]:
# do it
res = dock_obj.dock(mols,prot)
In [19]:
out= open("results.txt","w")
for i,r in enumerate(res):
print "%4d%10s%8s%8s%8s" %(i,r.formula, r.data['vina_affinity'], r.data['vina_rmsd_ub'], r.residues[0].name)
out.write("%4d%10s%8s%8s%8s\n" %(i,r.formula, r.data['vina_affinity'], r.data['vina_rmsd_ub'], r.residues[0].name))
In [49]:
for i,r in enumerate(res):
hbs = oddt.interactions.hbonds(prot,r)
stack= oddt.interactions.pi_stacking(prot,r)
phob = oddt.interactions.hydrophobic_contacts(prot,r)
print "%8s%8s%8s" %(len(hbs[0]),len(stack[0]),len(phob[0]))
#распечатать с результатами докинга
Отсортировали от лучшего заместителя к худшему, результат представлен в таблице.
45 C13H17NO6 -3.4 0 UNL 46 C13H17NO6 -3.3 5.162 UNL 47 C13H17NO6 -3.1 5.253 UNL 48 C13H17NO6 -3.1 2.288 UNL 49 C13H17NO6 -3.1 4.802 UNL 00 С8H15NO6 -2.9 0 UNL 09 C7H13NO7 -2.9 0 UNL 27 C7H15N2O6 -2.9 0 UNL 50 C13H17NO6 -2.9 5.453 UNL 36 C8H13NO8 -2.8 0 UNL 51 C13H17NO6 -2.8 9.345 UNL 52 C13H17NO6 -2.8 3.049 UNL 53 C13H17NO6 -2.8 8.249 UNL 18 C7H13NO6 -2.7 0 UNL 37 C8H13NO8 -2.7 3.224 UNL 01 C8H15NO6 -2.6 5.28 UNL 10 C7H13NO7 -2.6 5.136 UNL 11 C7H13NO7 -2.6 3.203 UNL 19 C7H13NO6 -2.6 3.588 UNL 20 C7H13NO6 -2.6 4.748 UNL 38 C8H13NO8 -2.6 4.939 UNL 39 C8H13NO8 -2.6 5.084 UNL 40 C8H13NO8 -2.6 6.975 UNL 02 C8H15NO6 -2.5 2.823 UNL 03 C8H15NO6 -2.5 5.231 UNL 04 C8H15NO6 -2.5 5.357 UNL 05 C8H15NO6 -2.5 3.209 UNL 06 C8H15NO6 -2.5 4.75 UNL 12 C7H13NO7 -2.5 3.039 UNL 13 C7H13NO7 -2.5 2.271 UNL 21 C7H13NO6 -2.5 2.498 UNL 28 C7H15N2O6 -2.5 5.169 UNL 41 C8H13NO8 -2.5 5.924 UNL 42 C8H13NO8 -2.5 3.506 UNL 43 C8H13NO8 -2.5 2.213 UNL 44 C8H13NO8 -2.5 3.844 UNL 07 C8H15NO6 -2.4 4.467 UNL 14 C7H13NO7 -2.4 1.946 UNL 29 C7H15N2O6 -2.4 3.211 UNL 30 C7H15N2O6 -2.4 1.879 UNL 08 C8H15NO6 -2.3 2.376 UNL 15 C7H13NO7 -2.3 4.97 UNL 16 C7H13NO7 -2.3 2.861 UNL 17 C7H13NO7 -2.3 4.424 UNL 22 C7H13NO6 -2.3 4.571 UNL 23 C7H13NO6 -2.3 2.481 UNL 24 C7H13NO6 -2.3 4.998 UNL 31 C7H15N2O6 -2.3 4.56 UNL 32 C7H15N2O6 -2.3 4.507 UNL 25 C7H13NO6 -2.2 4.572 UNL 33 C7H15N2O6 -2.2 4.787 UNL 34 C7H15N2O6 -2.2 4.266 UNL 35 C7H15N2O6 -2.2 2.119 UNL 26 C7H13NO6 -2.1 3.564 UNL
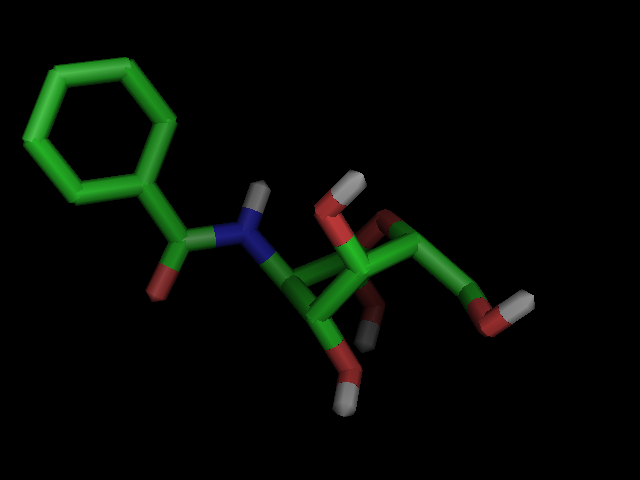
Лучшим заместителем оказался C13H17NO6 с лигандом - Ph. Ни в каком из соединений нет стакинг взаимодействий.
In [24]:
for i,r in enumerate(res):
r.write(filename='r%s.pdb' % i, format='pdb')
In [25]:
# на рисунке представлен лучший лиганд
import nglview
import ipywidgets
w1 = nglview.show_structure_file('r45.pdb')
w1