На предыдущем практикуме были выполнены все шаги, описанные в задании 7:
"Молекулярная динамика биологических молекул в GROMACS" Результаты были скачены с суперкомпьютера.
Изменение максимальной силы в ходе оптимизации геометрии:
начальное значение максимальной силы = Step 0 Fmax = 4.02438e+03;
конечное значение максимальной силы = Step 86 Fmax = 2.12381e+03.
Анализ
- Силовое поле используемое при построении топологии: amber99sb
- Заряд системы: был -10(ДНК отрицательно заряжена), стал 0, так как мы его нейтрализовали.
- Размер и форму ячейки: кубическая 5.01400 x 5.00700 x 5.26800
- Минимизация энергии:
- Алогритм минимизации энергии: integrator = l-bfgs; Goldfarb-Shanno (квази-Ньютоновский), метод аппроксимирует Гауссиановскую матрицу из предыдущих конфигураций
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий: cut-off ("двойное обрезание").
- Модель, которой описывался растворитель: implicit_solvent = No, то есть ЯВНЫЙ
- Утряска растворителя:
- Число шагов: nsteps = 10000
- Длина шага: delta_t = 0.001 (ps)
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий: pme (суммирование по Эвальду)
- Алгоритмы термостата и баростата: температура Berendsen, контроля давления не проиходило
- Основной расчёт МД:
- Время моделирования, количество процессоров, эффективность маштабирования.
- Число шагов: 5000000
- Длина шага: 0.002 ps
- Алгоритм интегратора: md
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий: pme и cutt-off
- Алгоритмы термостата и баростата: Berendsen
Любой анализ начинаем с визуального анализа движений молекул. При вопросе о выводе групп выбераем DNA.
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_pbc_1.pdb -skip 20 -pbc molОткрываем b_pbc_1.pdb в PyMol.
В ролике молекула мечется по экрану, пробуем:
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_fit_1.pdb -skip 20 -fit rot+transОткрываем dna_fit_1.pdb в PyMol.
ДНК уже не мечется по экрану, правда в какой-то момент кажется, что цепи почему-то расходятся, но если рассмотреть ДНК относительно введенной нами ячейки, то понятно, что молекула просто выходит за ее границы и появляется с другой стороны.

Мне кажется, что в состоянии 5 ДНК уже похожа на В-форму.
TITLE Protein in water t= 800.00000 REMARK THIS IS A SIMULATION BOX CRYST1 50.033 49.963 52.568 90.00 90.00 90.00 P 1 1 MODEL 5
Время моделирования 0,8 нс.
Определим средне-квадратичное отколнение в ходе моделирования. Так как у нас происходит конформационный переход сначала расчитаем отклонение в ходе всей симуляции относительно стартовой структуры.
g_rms -f dna_md.xtc -s dna_md.tpr -o rms_1
 И относительно каждой предидущей структуры на растоянии 400 кадров. Если ближе к концу закончился конформационный переход, то отколнение должно уменьшаться.
И относительно каждой предидущей структуры на растоянии 400 кадров. Если ближе к концу закончился конформационный переход, то отколнение должно уменьшаться.
g_rms -f dna_md.xtc -s dna_md.tpr -o rms_2 -prev 400

Графики выглядят почти одинакого, но можно заметить, что к концу временной шкалы во втором случае квадратичное отклонение, действительно, становится меньше.
Определим изменение гидрофобной и гидрофильной поверхности в ходе конформационного перехода.
g_sas -f dna_md.xtc -s dna_md.tpr -o sas_dna.xvg
В полученном файле второй и третий столбцы -- гидрофобная и гидрофильная посверхности соответственно.
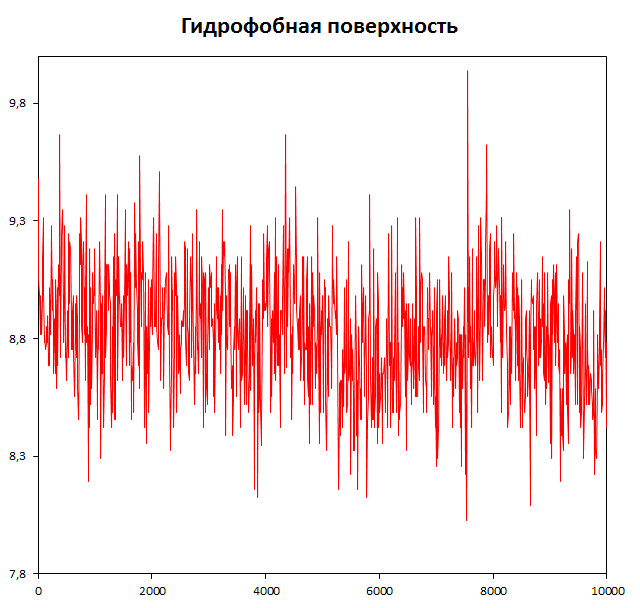

ДНК находится в воде. К концу временного промежутка гидрофобная поверхность незначительно снижается, видимо, это способствует конформационному переходу.
Традиционным анализом для ДНК является расчёт колчества образуемых водородных связей. Если мы будем исследовать связи между ДНК и ДНК, то это будут водородные связи между цепями ДНК. Для конца траектории:
g_hbond -f dna_md.xtc -s dna_md.tpr -num hbond_dna
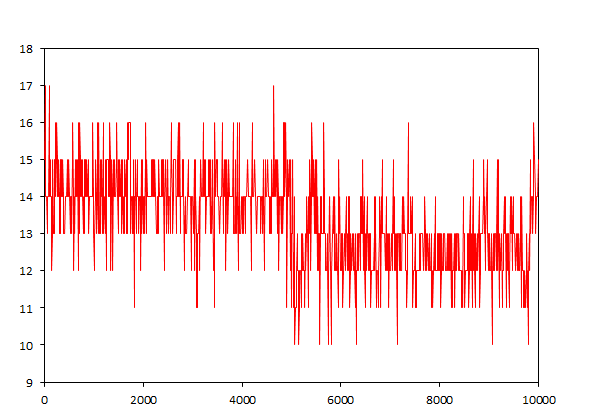
Вообще в каноническом дуплексе между цепями должно быть 14 связей, а тут мы видим значения от 12 до 15, что недалеко от 14. Правда имеются выбросы вплоть до 10 или 17, но, возможно, это связано с ошибками. Среднее значение колеблется между 13 и 14, и будем считать, что оно мало изменяется во времени.
Не менее интересно будет изучить количество вдородных связей ДНК-Вода
g_hbond -f dna_md.xtc -s dna_md.tpr -num hbond_dna_sol
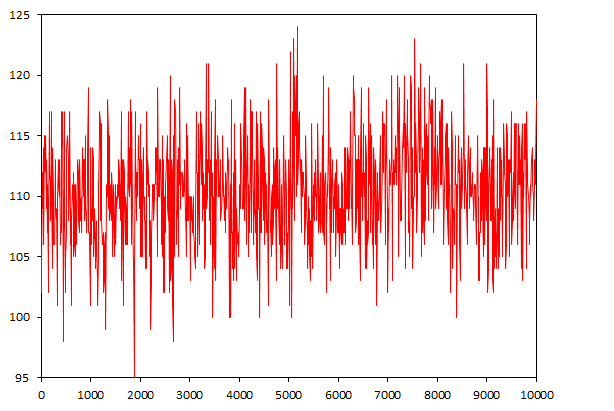
Количество водородных связей варьирует от 95 до 124 и вцелом незначительно изменяется с течением времени. Видимо, небольшие изменения в количестве водородных связей не мешают ДНК перейти из одной формы в другую.