Выравнивания последовательностей 3
Циклотиды
Циклотиды - это растительные белки, замкнутые в кольцо. От других циклических белков они отличаются бОльшим размером(в них около 30 ак, а например а-аманитин бледной поганки - 8 ак), наличием вторичной и третичной структур(например, в них есть дисульфидные связи). Благодаря кольцевой структуре, 3 дисульфидным связям и не большому размеру эти белки очень стабильны - не режутся протеазами и не денатурируют при высоких температурах. Предполагается, что эти белки участвуют в защите растений от патогенов.
Сравнение выравниваний muscule и ClustalW
Взятые белки:
- Rinorea virgata(6DHR)
- Chassalia parviflora(1BH4)
- Chassalia parviflora(2ERI)
- Viola odorata(2GJ0)
- Oldenlandia affinis(1KAL)
- Palicourea condensata(1R1F)
Исходное выравнивание в muscule
Исходное выравнивание в ClustalW
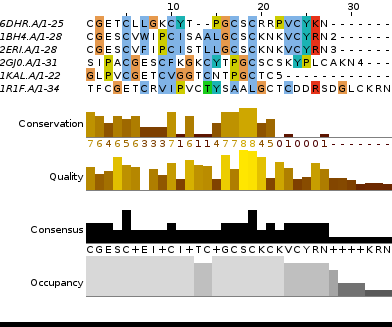
В белке 1KAL передняя часть не выравнивается с остальными, а кончается в выравнивании он раньше других. Скорей всего, из-за того, что белок циклический и был записан начиная с другой аминокислоты.
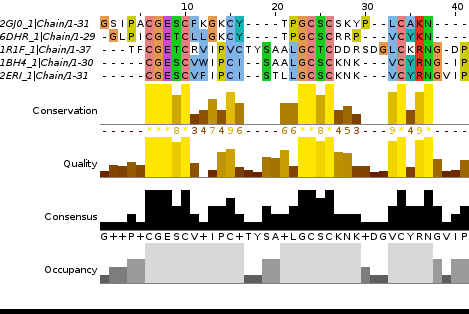

У первых двух последовательностей тоже есть такая проблема - фрагмент у них в начале похож на тот, который у остальных последовательностей в конце, но он не большой.
На обоих выравниваниях видно 3 консервативных блока: 6-16, 19(22)-26 и 33-38.
Три дисульфидных связи - специфическая особенность циклотидов. ClustalW выравнивает все 6 цистеинов правильно, а muscule - только 5.
Структурное выравнивание
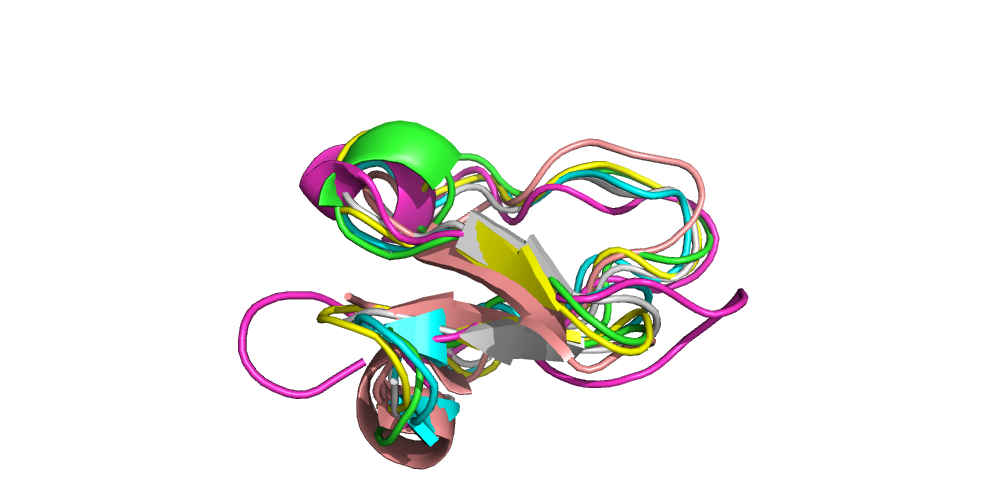
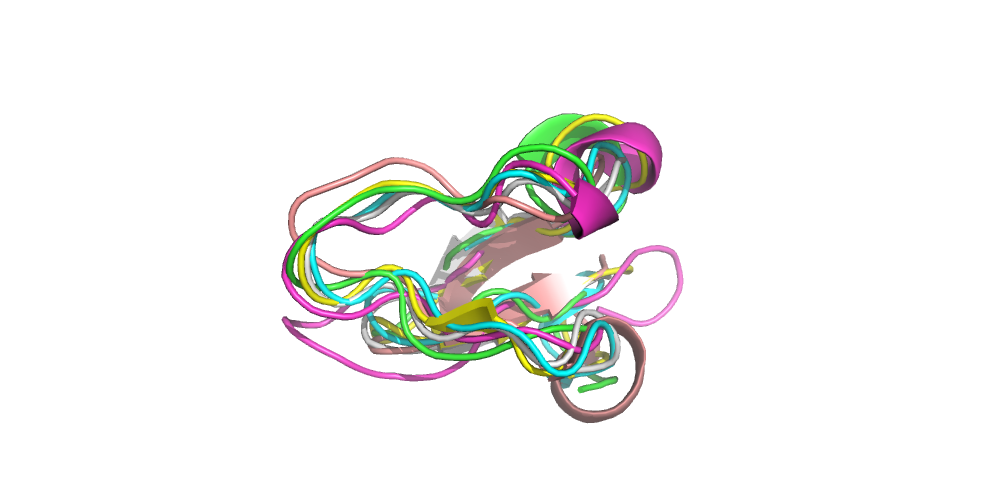
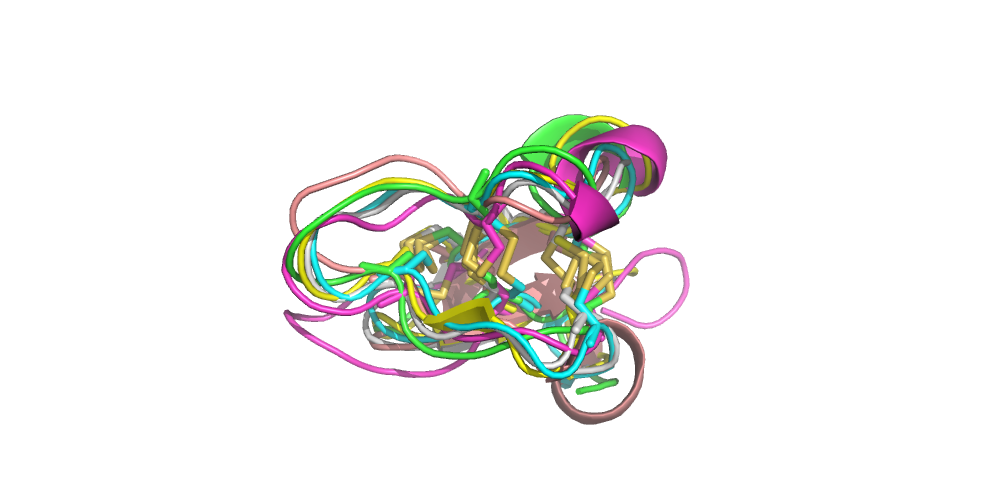
Структурное выравнивание вышло гораздо хуже, чем оба множественных - ни один из 6 цистеинов не выровнялся, блоки тоже не выделяются. Скорей всего, из-за того, что структурное выравнивание делалось попарно, а не множественным выравниванием. При этом по самим выровненным структурам в pymol хорошо видно, что белки гомологичны, а дисульфидные связи почти совпадают.
Описание работы программы ClustalW
Сначала программа выравнивает все последовательности попарно. Из этих выравниваний она получает матрицу расстояний между последовательностями. По ней строится направляющее дерево. В ранних версиях оно строилось по методу Neighbour-Joining, позже было заменено на UPGMA - он менее точный, но значительно более быстрый. После построения дерева попарные выравнивания выравниваются с другими последовательностями и друг другом.
Один из минусов этого алгоритма - невозможность "исправить ошибку", появившуюся в первых выравниваниях: например, если в одном из первых выравниваниях был открыт гэп, то он будет появляться в последующих выравниваниях.
Также алгоритм плохо работает на далнеродстственных последовательностях: значения в матрице расстояний оказываются большими и могут не отражать степень родства.