| Главная страница | Обучение | Обо мне | Ссылки | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Банки нуклеотидных последовательностей: Часть 2 Поиск по сходству | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Банки нуклеотидных последовательностей. Часть 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Задание 4 (1). В практикуме 6 по данным хроматограмм была получена нуклеотидная последовательность WS2980_H3_F_A03_final. Для того чтобы понять, к какому гену и какому организму она относится, по ней был запущен BLASTN. Параметры поиска:
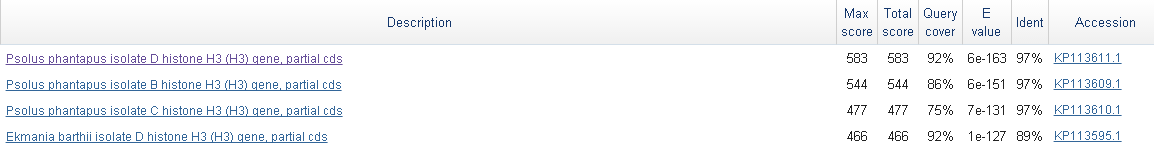
Результаты поиска Было найдено 20 000 схожих последовательностей, у самой лучшей находки E-value 6e-163, у худшей - 3e-66 с процентом идентичности 76% (см. Рис.1), то есть ее в целом нельзя назвать плохой. К сожалению, 20 000 - максимальное возможное число находок, которое мы можем получить при выдаче (ограничение, введенное параметром поиска).
Если бы можно было выводить большее количество последовательностей, похожих на введенную, наверняка нашлось бы еще много последовательностей. При этом все находки относятся к генам белка гистона H3, следовательно, последовательность, полученная по хроматограмме в предыдущем практикуме, также является частью гена гистона H3. В этом случае такое большое число достоверных находок объясняется тем, что аминокислотная (а значит, и нуклеотидная) последовательность гистонов является консервативной и практически не различается у эукариотических организмов из разных таксонов.
Ниже (в Таблице 1) представлены данные по нескольким лучшим находкам. Выравнивания входной последовательности и этих находок представлены в проекте Jal-view. Исходная посл-ть везде обозначена как sequence, а находки - в соответствии с их ID. Таблица 1. Лучшие находки, полученные в результате поиска последовательности WS2980_H3_F_A03_final.fa в blastn.
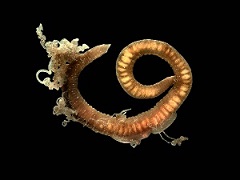
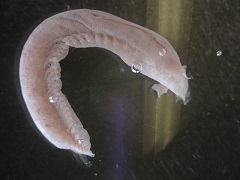
На рис. 2 представлены некоторые организмы из этой таблицы:
Второй ряд, слева направо: Thoracophelia mucronata, Ophelia limacina, Chiridota sp. Таксономия данных организмов: рис. 3.
Было построено выравнивание последовательности WS2980_H3_F_A03_final.fa, полученной по хроматограмме, с выбранными находками, представленными в Таблице 1:  По таксономическим данным можно сделать вывод, что искомый ген принадлежит либо голотуриям (вторичноротые, иглокожие), либо многощетинковым червям (первичноротые). Однако, учитывая процент идентичных колонок и общий вес (score) выравниваний, можно предположить, что скорее всего исходная последовательность является частью гена гистона H3 организмов, принадлежащих классу Holothuroidea, а точнее - организму Psolus phantapus. Уровень сходства посл-ти WS2980_H3_F_A03_final.fa и части гена histone H3 организма Psolus phantapus: 3 замены на 100 пн. Сходство посл-ти с геном того же белка из организма Ekmania barthii: 11 замен на 100 пн. Сходство посл-ти с геном гистона H3 организма Ophelia limacina: 13 замен на 100 пн. Поиск по сходству. Часть I
| Задание 2. В этом задании нужно было сравнить результаты поиска нуклеотидной последовательности с помощью трех разных алгоритмов blastn. Была взята последовательность WS2980_H3_F_A03_final.fa. Были заданы следующие параметры поиска:
Результаты Результаты поиска данной нуклеотидной последовательности с помощью трех алгоритмов blastn приведены в Таблице 2. Таблица 2. Результаты поиска посл-ти WS2980_H3_F_A03_final.fa с помощью различных алгоритмов blastn.
К сожалению, мне не удалось подобрать такой таксон, чтобы количество находок было от 100 до 1000, так как если брать таксон, например, на порядок выше, то на выходе получается несколько тысяч находок. Однако разброс процента сходства полученных тремя разными алгоритмами находок достаточно хороший - от 97% до 75%. Из Таблицы 2 видно, что алгоритмы discontiguous megablast и megablast отсекают находки с очень высоким E-value. На рис. 4 представлен фрагмент списка тех результатов поиска алгоритмом blastn, которые не попали в результаты поиска по алгоритмам discontiguous megablast и megablast. Они имеют очень низкий query cover и процент сходства и скорее всего не являются гомологами искомой последовательности (это подтверждается даже их названиями). Их E-value оказался слишком высоким для этих более точных алгоритмов.
Что касается различий в результатах поиска по discontiguous megablast и megablast, то в список находок megablast попадают только те последовательности, которые имеют участки полного сходства длиной более 28 нуклеотидов (так как именно эта длина слова используется для megablast), а следовательно, у них должен быть высокий query cover.
Таких последовательностей довольно много, например, приведенные на Рис.5.
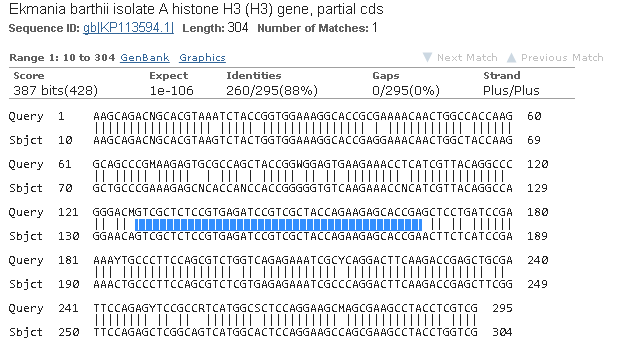
Из приведенных на Рис. 5 находок в список находок megablast попала только первая (Ekmania barthii isolate A histone H3 (H3) gene, partial cds), так как в ее выравнивании с искомой последовательностью присутствует участок совпадения длиннее 28 нуклеотидов (см. Рис.6).
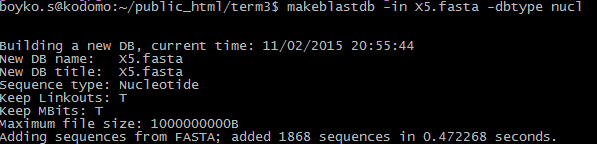
Благодаря такому длинному совпадающему фрагменту процент сходства последовательности гена гистона H3 Ekmania barthii (88%) выше, чем у последовательностей с близким E-value. При этом, однако, у этой находки query cover ниже (за счет того, что хорошо выравнивается лишь часть гена). Таким образом, полученные результаты показывают разницу между тремя алгоритмами blastn. Только megablast позволяет находить наиболее близких и достоверных гомологов для искомых последовательностей, discontiguous megablast позволяет находить гомологов, которые уже дивергировали и имеют какие-то схожие фрагменты и между ними небольшие различающиеся участки. Blastn (somewhat similar sequences) находит наиболее широкий круг последовательностей, многие из которых являются слишком далекими гомологами или могут не быть гомологами вообще. Задание 3.2 В этом задании нужно было найти гомологов нескольких белков в геноме организма X5 (Amoboaphelidium). Были взяты следующие белки: HS71B_MOUSE, RPB1_HUMAN, TERT_MOUSE, DPOD2_BOVIN, RS5_MOUSE. Поиск был произведен с помощью локального tblastn (пакет BLAST+, установленный на компьютер). Сначала была создана локальная база данных. B качестве такой базы был взят файл сборки генома Х5 X5.fasta. Это было сделано с помощью программы makeblastdb (показано на скриншоте).
Также аминокислотные последовательности перечисленных выше белков были скачаны и объединены в общий файл proteins.fasta. Это представляется более удобным, так как программа tblasn может принимать на вход файл с несколькими последовательностями, а на выходе выдает файл с результатами поиска для каждой последовательности, и использование общего входного файла экономит время. Затем была запущена программа tblastn с параметрами, приведенными на скриншоте ниже.
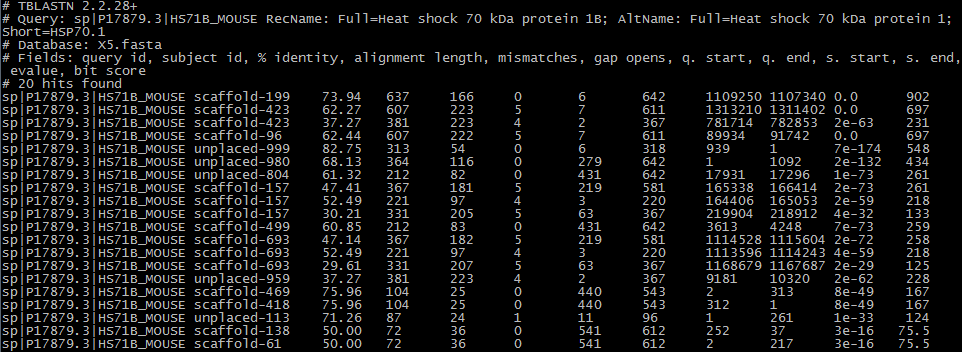
Далее будут рассмотрены результаты поиска tblastn по геному X5 гомологов каждого белка по отдельности. Данные взяты из файла, созданного программой tblastn, tblastn.out. 1. HS71B_MOUSE (AC:P17879) - белок теплового шока, шаперон. Он (совместно с другими шаперонами) участвует в процессе фолдинга новообразующихся белков, предотвращает нежелательное сворачивание белков в ходе их посттрансляционного транспорта в пластиды и митохондрии. Кроме этого, он играет важную роль в убиквитинировании и деградации белков. Все это возможно благодаря способности белков семейства Hsp70 распознавать ненативную конформацию того или иного белка. . Длина белка - 642 аминокислотных остатка. Tblastn нашел 20 нуклеотидных последовательностей в сборке Х5, которые теоретически могут быть гомологами данного белка. Таблица с полученными находками - см. Рис.7.
На мой взгляд, самыми хорошими находками среди этих 20 являются 4. Они подробно описаны в Таблице 3.
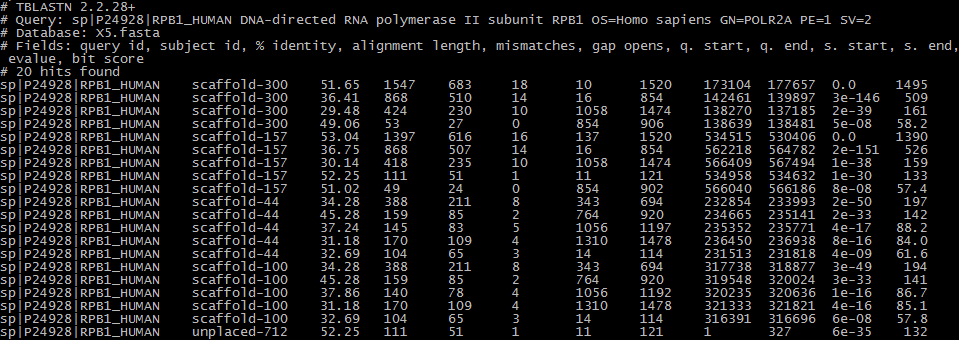
Как видно из Таблицы 3, эти четыре находки хороши каждая по-своему. У них у всех E-value существенно меньше, чем у остальных находок. Однако наилучшей, по моему мнению, следует считать самую первую находку, так как, во-первых, она покрывает HSP71B практически полностью, при этом имеет очень высокий вес и E-value 0.0. У следующих двух находок так же E-value равен 0.0, но процент идентичности ниже на ~10% и соответственно ниже score. Четвертая находка может показаться наиболее удачной, если смотреть только на процент идентичности и E-value, однако следует заметить, что она покрывает меньше половины искомой последовательности, а значит, не может однозначно являться гомологом белка HSP71B. Таким образом, наиболее вероятно, что гомолог белка HS71B_MOUSE в геноме X5 (Amoeboaphelidium) принадлежит скэффолду сборки scaffold-199. Ее параметры приведены в Таблице 2. 2. RPB1_HUMAN (AC: P24928) - управляемая ДНК субъединица RPB1 РНК-полимеразы II. Она содержит карбоксиконцевой домен, в который входят до 52 повторов паттерна YSPTSPS. Этот домен необходим для связывания с белками, которые инициируют полимеразную активность. RPB1-субъединица входит в ядро РНК-полимеразы, являясь своеобразным зажимом, который при перемещении открывает и закрывает щель, через которую проходит транскрибируемая ДНК. Длина белка - 1970 амк остатков. Tblastn выдал 20 находок из сборки генома Х5 для искомой последовательности. Они приведены на рис. 8.
Из представленных находок 2 бросаются в глаза своим явным отличием (в сторону своей достоверности) от остальных: 1-ая (scaffold-300) и 5-я (scaffold-157). Их характеристики приведены в Таблице 4. Таблица 4. Характеристика лучших находок гомологов для последовательности белка RPB1_HUMAN.
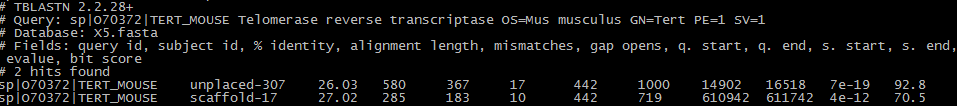
Можно считать, что в scaffold-300 входит предполагаемый гомолог PPB1-субъединицы РНК-полимеразы, так как ее показатели в целом лучше, чем для второй находки, а именно query cover выше, чем у scaffold-157 при близких значениях процентов сходства. 3. TERT_MOUSE (AC: O70372) - теломераза, обратная транскриптаза, компенсирующая укорочение теломер, происходящее при делении клетки в ее жизненном цикле. В здоровых соматических клетках имеет низкую активность, а в раковых или стволовых - напротив, высокую. Катализирует РНК-зависимое присоединение короткой последовательности 5'-TTAGGG-3' к 3'-концу хромосомы. Длина белка - 1122 амк остатка. Программа tblastn выдала всего 2 находки гомолога последовательности данного белка в геноме X5. Они показаны на рис. 9.
Результаты находок продублированы в Таблице 5 в более явном виде. Таблица 5. Характеристика лучших находок гомологов для последовательности белка TERT_MOUSE.
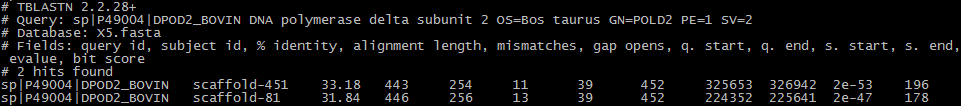
Результат на первым взгляд не показался мне хорошим, поэтому я попробовала провести поиск по геному X5 с порогом E-value = 1. Это позволило добавить к двум находкам еще одну со следующими характеристиками: 31.82% identity, query cover 5,44%, E-value 0.27, score 35.0. На основании этих чисел можно заключить, что новая находка является менее достоверной, чем полученные ранее, так как хоть она и имеет более высокий процент сходства, он достигается за счет очень малого покрытия последовательности, а значит, не может свидетельствовать об однозначной гомологии. Поэтому, скорее всего, лучшая из двух находок, а именно unplaced-307 включает в себя какую-то часть гена белка TERT_MOUSE. 4. DPOD2_BOVIN (AC: P49004) - субъединица 2 ДНК-полимеразы δ. Эта ДНК-полимераза входит в семейство В ДНК-полимераз и осуществляет синтез лидирущей цепи в ходе репликации. Также обладает 3'-5'-эндонуклеазной активностью, благодаря которой может выполнять корректорскую функцию в ходе синтеза ДНК. Белок DPOD2_BOVIN является малой субъединицей ДНК-полимеразы (~50кДа), которая служит фактором димеризации репликативных холоферментов. Длина белка - 469 амк остатков. Ниже (на рис. 10) приведен результат поиска последовательности данного белка по геному X5. Всего получено 2 находки.
Эти 2 находки довольно похожи друг на друга по своим характеристикам (представлены ниже в Таблице 6, их query cover одинаков, по таблице на Рис. 10 видно, что различие между находками всего в 2 гэпа (для второй находки). Поэтому возможно, что гомолог белка DPOD2_BOVIN представлен двумя копиями гена, попавшими в разные скэффолды. Таблица 6. Характеристика лучших находок гомологов для последовательности белка DPOD2_BOVIN.
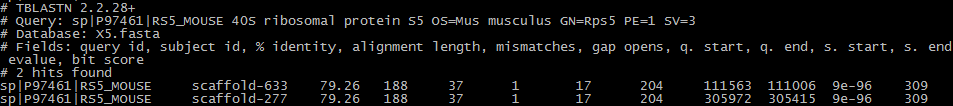
5. RS5_MOUSE (AC: P97461) - рибосомальный белок S5 эукариот, входящий в состав субъединицы 40S. Является достаточно консервативным, как и все рибосомальные белки. Длина белка - 204 амк остатка. Результат выдачи tblastn показан ниже, на рис. 11.
И в этом поиске было получено всего 2 находки гомологов, демонстрирующие довольно высокую достоверность. Их характеристики представлены ниже в Таблице 7. Таблица 7. Характеристика лучших находок гомологов для последовательности белка RS5_MOUSE.
Как можно заметить из Таблицы 7, находки являются совершенно одинаковыми с точки зрения своих характеристик и отличаются лишь тем, что находятся в разных скэффолдах. Так что можно сказать, что гомолог белка RS5_MOUSE находится и в scaffold-633, и в scaffold-277, и он представлен в геноме X5 в двух одинаковых копиях. Поиск по сходству. Часть II
| Задание 4. В этом задании нужно было создать классификацию родственных вирусов по сходству их последовательностей. Был взят Rabies virus, вирус бешенства, изучавшийся в первом семестре, а конкретно - 5 разных штаммов этого вида. Это следующие штаммы:
Затем был создан файл rv_full.fasta, в котором объединены все последовательности геномов пяти выбранных штаммов (в том же порядке, что и в списке выше). Далее использовался локальный blast:
После того, как была получена таблица находок tblastx, ее необходимо было "почистить" с помощью python-скрипта. Так как исходно были взяты последовательности штаммов одного и того же вида, то можно предположить, что они должны быть весьма похожи друг на друга. Поэтому параметры для скрипта были немного изменены:
В результате была получена таблица hits1.xls. Была проведена последовательная сортировка этой таблицы: сначала по query_id (чтобы увидеть для каждого штамма его ближайшего родственника), затем по identityи по aln_len, то есть по длине выравнивания. Я посчитала, что этот параметр также важен, так как штаммы одного вида близки между собой, длинное выравнивание с высоким процентом идентичности могло бы быть доводом в пользу их родства. Параметр E-value в такой ситуации, напротив, роли не играет - у подавляющего большинства выравниваний в этой таблице он равен 0.0. Отсортированная таблица есть в файле hits1_sorted.xls.
© Alexandra Boyko, 2014. Faculty of Bioengineering and Bioinformatics, MSU. | | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||