Анализ результатов моделирования сборки бислоя
Файлы моделирования были скачаны с суперкомпьютера "Ломоносов" в папку на kodomo. Все дальнейшие команды вводились прямо в терминале, в отчете ниже они дублируются в блоках с %%bash.
В терминале на кодомо была запущена команда ниже и выбрана группа 2 (DРРС). Полученный pdb-файл был просмотрен в PyMol, визуализация показана на Рис. 1.
%%bash
trjconv -f b_md.xtc -s b_md.tpr -o b_pbc_1.pdb -skip 20 -pbc mol
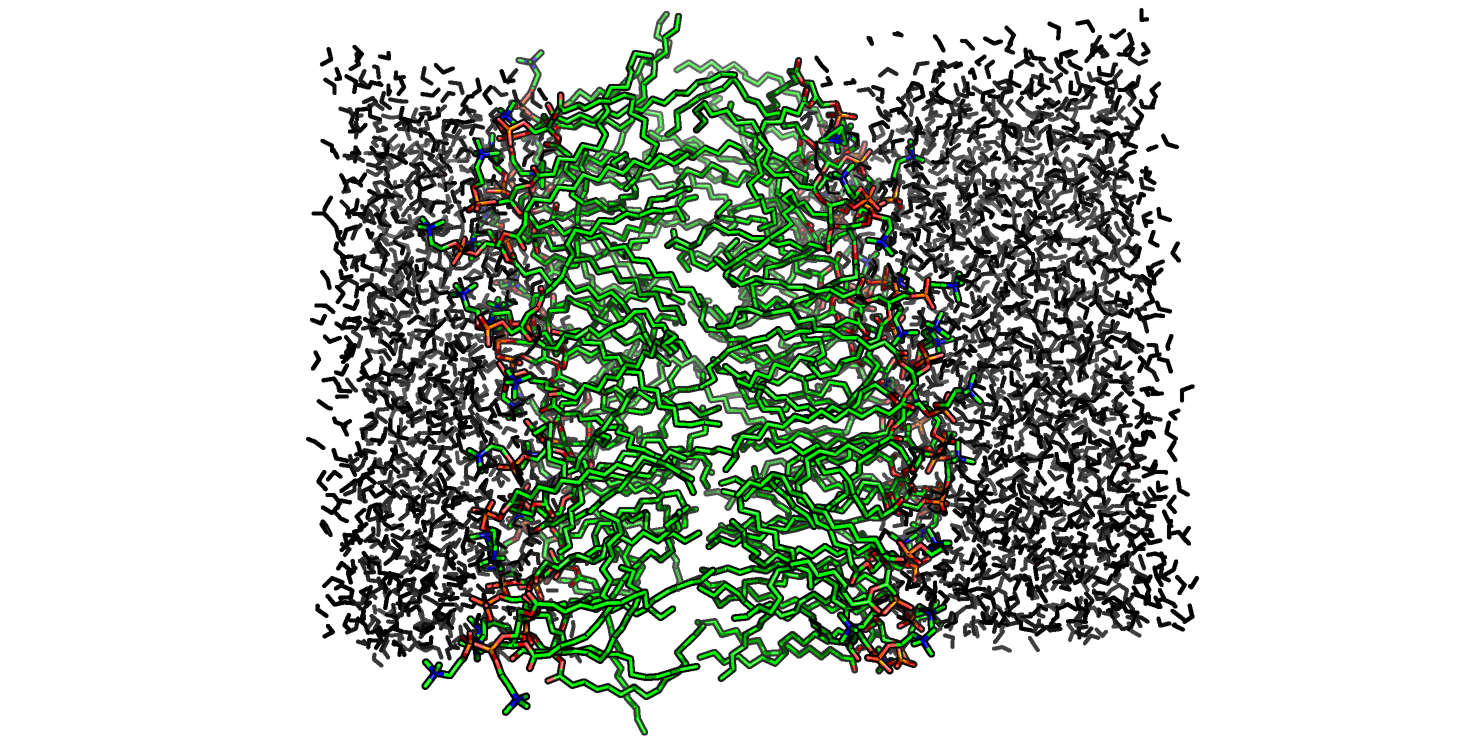
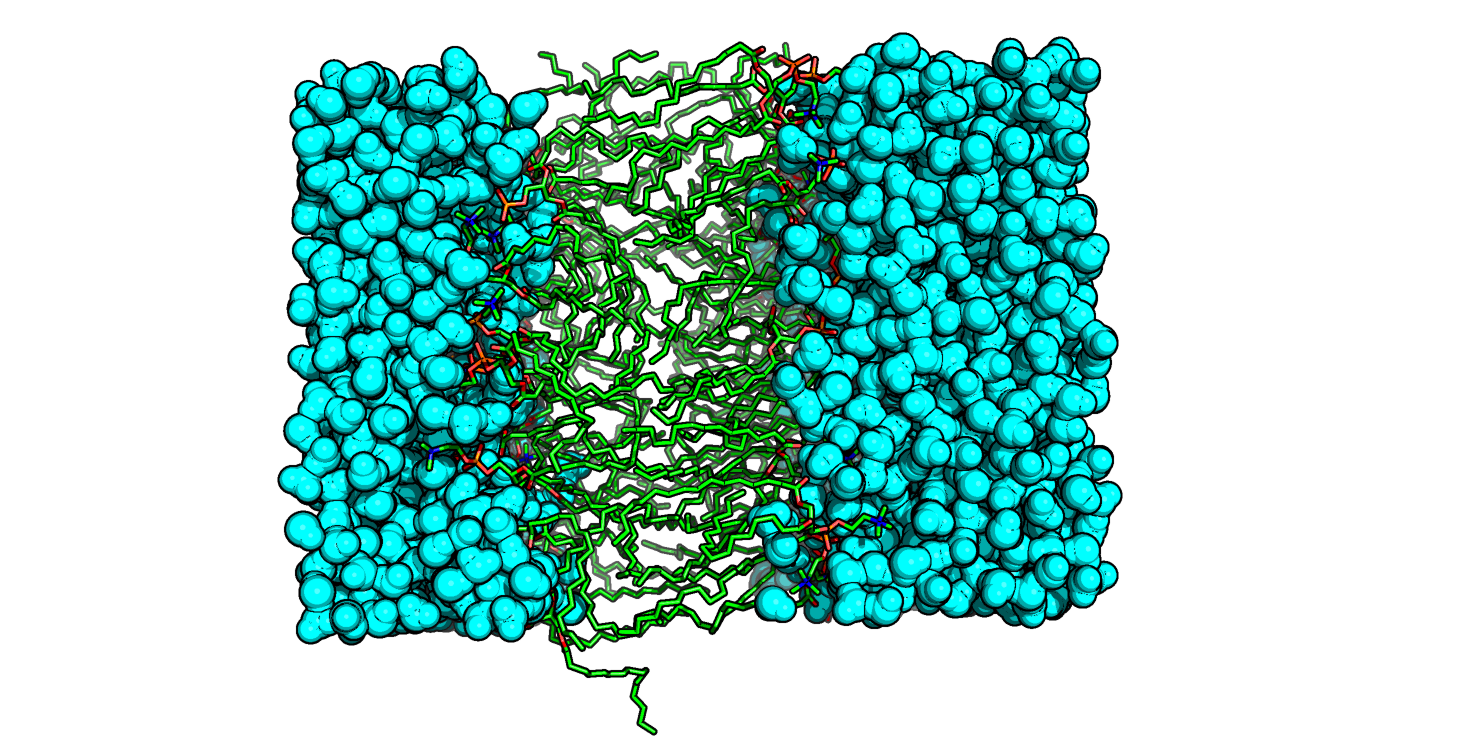
Билипидоподобная структура возникает уже на 20-ой модели (на 9500-ой пикосекунде), а до этого (модели с 7-ой) формируется мицеллоподобная структура. Более точно это можно оценить, если выбрать System, а не DPPC в предыдущей команде. Как раз на модели 20 молекулы воды не встречаются в гидрофобной части слоя (Рис. 2). Правда, сам билипид еще "мотает", гидрофобные хвосты не лежат так упорядоченно, как лежат на последней модели (Рис. 1).
Далее нужно было оценить зависимость площади поверхности одного липида от шага моделирования (времени).
Нормалью к поверхности бислоя является ось Х.
%%bash
g_traj -f b_md.xtc -s b_md.tpr -ob box_1.xvg
%matplotlib inline
import numpy as np
import matplotlib.pyplot as plt
from scipy import optimize
#подготовим данные для графика
time = []
square = []
with open("box_1.xvg", 'r') as boxfile:
lines=boxfile.readlines()
for each_line in lines:
if each_line.startswith("@") or each_line.startswith("#"):
continue
else:
lines_list = each_line.split()
time.append(lines_list[0])
square.append(float(lines_list[2])*float(lines_list[3])/32)
plt.plot(time, square, c='gold',alpha=1)
plt.ylabel('S')
plt.xlabel('Time, ps')
plt.show()

Площадь поверхности одного липида падает, что крайне логично. Небольшое увеличение площади на последних пикосекундах (моделях) объясняется подвижностью и небольшим изгибанием билипидного слоя.
Теперь изучим изменение гидрофобной и гидрофильной поверхности в ходе сборки билипидного слоя.
%%bash
g_sas -f b_md.xtc -s b_md.tpr -o sas_b.xvg
time = []
hphobic = []
hphylic = []
with open("sas_b.xvg", 'r') as sasfile:
lines=sasfile.readlines()
for each_line in lines:
if each_line.startswith("@") or each_line.startswith("#"):
continue
else:
lines_list = each_line.split()
time.append(lines_list[0])
hphobic.append(lines_list[1])
hphylic.append(lines_list[2])
plt.plot(time, hphobic, c='navy',alpha=1)
plt.plot(time, hphylic, c='pink',alpha=1)
plt.legend(['hydrophobic SAS', 'hydrophilic SAS'], loc='upper right')
plt.ylabel('SAS')
plt.xlabel('Time, ps')
plt.show()

Наблюдается резкое снижение гидрофобной поверхности, доступной растворителю, в самом начале сборки (липиды формируют "рыхлую" мицеллу, Рис. 3), потом уже в ходе преобразования в билипид и его утряске уменьшение площади происходит более постепенно (скорее даже практически не происходит, начиная с 20000 пикосекунд). Гидрофильная поверхность логичным образом более доступна полярному растворителю (кривая ее площади выше, чем кривая для гидрофобной поверхности). При этом в начале она самая большая за счет того, что липиды неупорядочены или формируют мицеллу, а сама мицелла довольно рыхлая и вода может легко облеплять полярную головку липида. Потом в ходе уплощения билипидного слоя площадь гидрофильной поверхности, доступная растворителю, немного снижается, потому что липиды сближаются между собой и вода уже не может легко проникать между полярными головками.
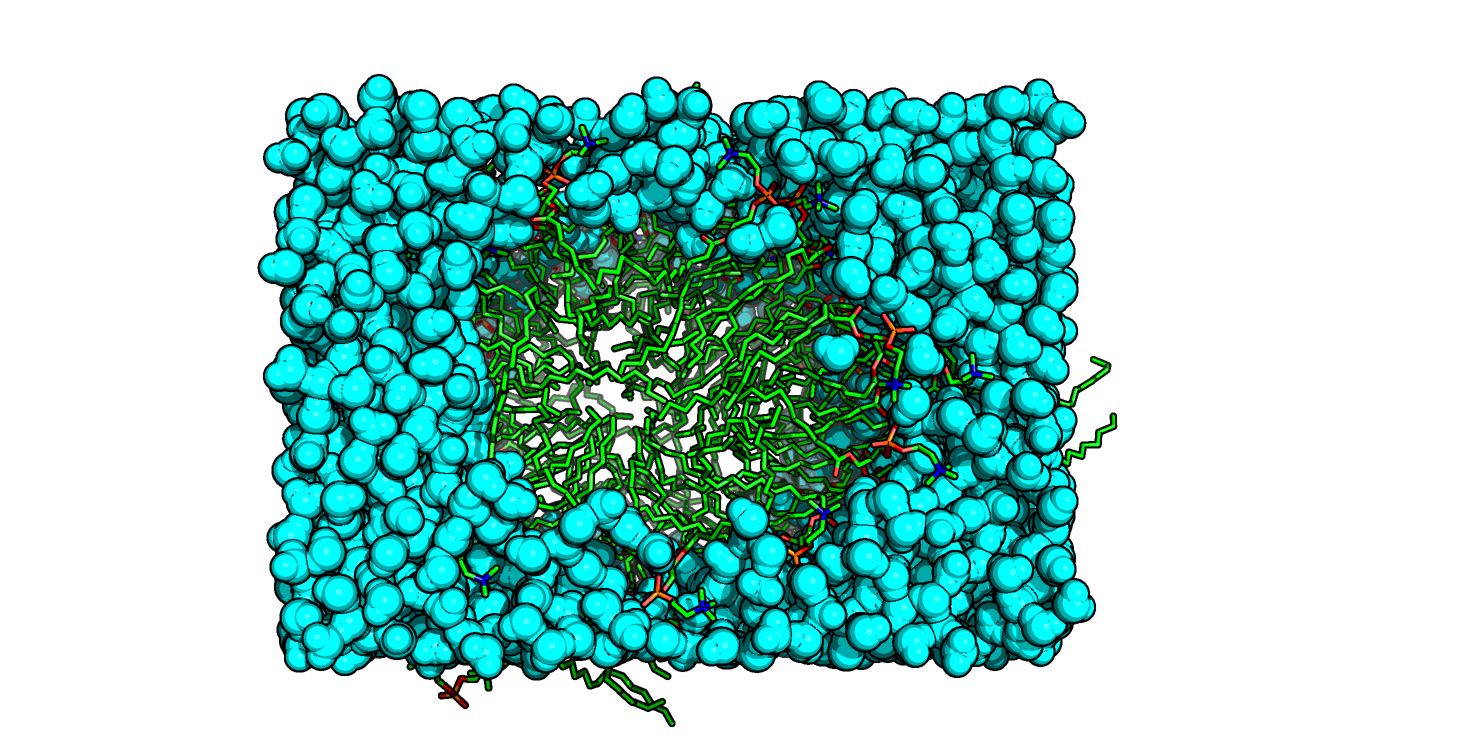
Оценим фазовое состояние бифильных молекул липидов (определим меру порядка).
Для этого скачаем специальный индекс-файл sn1.ndx.
! wget http://kodomo.cmm.msu.ru/~golovin/bilayer/sn1.ndx
Как уже было сказано выше, нормалью к поверхности бислоя является ось Х.
%%bash
g_order -s b_md -f b_md.xtc -o ord_end.xvg -n sn1.ndx -b 45000 -d X
g_order -s b_md -f b_md.xtc -o ord_start.xvg -n sn1.ndx -e 5000 -d X
После получили файлы deuter_start.svg и deuter_end.svg. В них для каждого атома указан параметр Scd - мера порядка, рассчитываемая по формуле:
{kind=link}
{kind=link}
##подгтовка данных для графика в начале траектории
atom_n = []
scd_start = []
with open("deuter_start.xvg", 'r') as deuterfile:
lines = deuterfile.readlines()
for each_line in lines:
if each_line.startswith("@") or each_line.startswith("#"):
continue
else:
lines_list = each_line.split()
atom_n.append(lines_list[0])
scd_start.append(lines_list[1])
##подгтовка данных для графика в конце траектории
atom_n = []
scd_end = []
with open("deuter_end.xvg", 'r') as deuterfile:
lines = deuterfile.readlines()
for each_line in lines:
if each_line.startswith("@") or each_line.startswith("#"):
continue
else:
lines_list = each_line.split()
atom_n.append(lines_list[0])
scd_end.append(lines_list[1])
plt.plot(atom_n, scd_start, c='olive', alpha=1, linewidth=3.0)
plt.plot(atom_n, scd_end, c='purple', alpha=1, linewidth=3.0)
plt.legend(['Scd (start)', 'Scd (end)'], loc='upper right')
plt.ylabel('Scd')
plt.xlabel('Atom number (C34 - C50)')
plt.show()

При идеальной упорядоченности Scd = 1. Здесь же мы наблюдаем довольно низкую степень упорядоченности в самом начале сборки (оливковая кривая на графике выше) и более высокую - на последних стадиях сборки липидного бислоя (фиолетовая кривая). При этом Scd в конце падает от меньшего номера атома к большему. Чем больше номер атома, тем ближе он к концу и тем более возможна неупорядоченность хвоста липида в гидрофобном слое.
Ниже приведена информация про процесс моделирования сборки липидного бислоя.
- Силовое поле, используемое при построении топологии: ffgmx (модифицированное силовое поле GROMOS87).
- Заряд системы: 0, система нейтральна.
- Форма и размер ячейки: объем 160.70 nm3 (6.260 nm х 4.443 nm х 5.778 nm), форма ячейки - параллелепипед, все углы 90 градусов.
- Алгоритм минимизации: l-bfgs
- Алгоритм расчета электростатики и Ван-дер-Ваальсовых взаимодействий: coulombtype = Cut-off, vdw-type = Cut-off
- Число шагов: 1000
- Длина шага: 0.002
- Алгоритм расчета электростатики и Ван-дер-Ваальсовых взаимодействий: pme, vdw-type = Cut-off
- Алгоритм термостата и баростата: Tcoupl = Berendsen, Pcoupl = no
- Число шагов: 10000000
- Длина шага: 0.005 ps
- Алгоритм интегратора: md (A leap-frog algorithm for integrating Newton’s equations of motion)
- Алгоритм расчета электростатики и Ван-дер-Ваальсовых взаимодействий: pme (Fast smooth Particle-Mesh Ewald (SPME) electrostatics), vdw-type = cut-off
- Алгоритмы термостата и баростата: Tcoupl = V-rescale, Pcoupl = Berendsen
Минимизация энергии:
Модель, которой описывается растворитель: flexspc
Утряска растворителя:
Основной расчет молекулярной динамики:
P.S. Я честно сделала весь прак до 16.05, а после 16.05 только поправила (а не дописала) текст в некоторых местах.