Гомологичное моделирование комплекса белка с лигандом
В этом практикуме будет смоделирован комплекс лизоцима с лигандом.
import sys
sys.path.append('/usr/lib/modeller9v7/modlib/')
sys.path.append('/usr/lib/modeller9v7/lib/x86_64-intel8/python2.5/')
import modeller
import _modeller
import modeller.automodel
env=modeller.environ()
env.io.hetatm=True
Для моделирования с лигандом был взят лизоцим дальневосточной жабы Bufo gargarizans andrewsi (LYS_BUFGA). В качестве образца будем использовать известную структуру лизоцима форели.
! wget http://www.pdb.org/pdb/files/1lmp.pdb
--2018-05-12 16:31:34-- http://www.pdb.org/pdb/files/1lmp.pdb
Resolving www.pdb.org... 128.6.244.52
Connecting to www.pdb.org|128.6.244.52|:80... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: https://www.rcsb.org/pdb/files/1lmp.pdb [following]
--2018-05-12 16:31:34-- https://www.rcsb.org/pdb/files/1lmp.pdb
Resolving www.rcsb.org... 132.249.213.190
Connecting to www.rcsb.org|132.249.213.190|:443... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: http://files.rcsb.org/view/1lmp.pdb [following]
--2018-05-12 16:31:35-- http://files.rcsb.org/view/1lmp.pdb
Resolving files.rcsb.org... 132.249.213.141
Connecting to files.rcsb.org|132.249.213.141|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: unspecified [text/plain]
Saving to: `1lmp.pdb.2'
[ <=> ] 130,410 127K/s in 1.0s
2018-05-12 16:31:37 (127 KB/s) - `1lmp.pdb.2' saved [130410]
! wget http://www.uniprot.org/uniprot/P85045.fasta
--2018-05-11 21:26:55-- http://www.uniprot.org/uniprot/P85045.fasta Resolving www.uniprot.org... 128.175.240.211, 193.62.193.81, 18.188.143.193, ... Connecting to www.uniprot.org|128.175.240.211|:80... connected. HTTP request sent, awaiting response... 200 OK Length: 240 [text/plain] Saving to: `P85045.fasta' 100%[======================================>] 240 --.-K/s in 0s 2018-05-11 21:26:55 (31.8 MB/s) - `P85045.fasta' saved [240/240]
alignm=modeller.alignment(env)
alignm.append(file='P85045.fasta', align_codes='all',alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
## есть смысл поправить идентификаторы
alignm[0].code = 'BUFGA'
alignm[1].code = '1LMP'
read_pd_459W> Residue type NAG not recognized. 'automodel' model building
will treat this residue as a rigid body.
To use real parameters, add the residue type to ${LIB}/restyp.lib,
its topology to ${LIB}/top_*.lib, and suitable forcefield
parameters to ${LIB}/par.lib.
read_pd_459W> Residue type NDG not recognized. 'automodel' model building
will treat this residue as a rigid body.
To use real parameters, add the residue type to ${LIB}/restyp.lib,
its topology to ${LIB}/top_*.lib, and suitable forcefield
parameters to ${LIB}/par.lib.
alignm.salign()
alignm.write(file='BUFGA.ali', alignment_format='PIR')
SALIGN_____> adding the next group to the alignment; iteration 1
Посмотрим выравнивание
! cat BUFGA.ali
>P1;BUFGA sequence:: : : : :::-1.00:-1.00 SGKYISWEDSCSYLQLQKYERCELAKALKKGGLADFKGYSLENWICTAFHESGYNTASTNYNPPDKSTDYGIFQI NSRWWCNDYKTPRSKNTCNIDCKVLLGDDISPAIKCAKRVVSDPNGMGAWVAWKKYCKGKNLSQWTQGCKL---* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 ----------------KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNT-DGSTDYGIFQI NSRYWCDDGRTPGAKNVCGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV...*
Тут лиганда нет (это три точки в конце последовательности). Добавим лиганд и проверим еще раз выравнивание.
with open("BUFGA.ali", 'r') as ali:
seq = ali.read().split("*")
seq[0] = seq[0][:-2] + "..."
seq[1] = seq[1][:-3] + "-..."
new = "*".join(seq)
with open("BUFGA_lig.ali", 'w') as out:
out.write(new)
! cat BUFGA_lig.ali
>P1;BUFGA sequence:: : : : :::-1.00:-1.00 SGKYISWEDSCSYLQLQKYERCELAKALKKGGLADFKGYSLENWICTAFHESGYNTASTNYNPPDKSTDYGIFQI NSRWWCNDYKTPRSKNTCNIDCKVLLGDDISPAIKCAKRVVSDPNGMGAWVAWKKYCKGKNLSQWTQGCKL-...* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 ----------------KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNT-DGSTDYGIFQI NSRYWCDDGRTPGAKNVCGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV-...*
## Выбираем объект для моделирования
s = alignm[0]
pdb = alignm[1]
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='BUFGA_lig.ali', knowns= pdb.code , sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 3
a.make()
BUFGA 1LMP
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
END OF TABLE
read_to_681_> topology.submodel read from topology file: 3
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 10 146
atom names : C +N
atom indices : 1164 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 10 146
atom names : C CA +N O
atom indices : 1164 1159 0 1165
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 4
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 14445 13409
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 149
Number of all, selected real atoms : 1209 1209
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 13409 13409
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2652
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1045.6554
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1197 0 0 0.007 0.007 14.018 1.000
2 Bond angle potential : 1618 0 9 2.318 2.318 173.69 1.000
3 Stereochemical cosine torsion poten: 814 0 31 47.518 47.518 286.37 1.000
4 Stereochemical improper torsion pot: 502 0 1 1.489 1.489 24.560 1.000
5 Soft-sphere overlap restraints : 2652 1 2 0.007 0.007 16.516 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2405 0 0 0.130 0.130 37.730 1.000
10 Distance restraints 2 (N-O) : 2564 2 12 0.233 0.233 134.51 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 145 1 7 5.364 5.364 49.192 1.000
14 Sidechain Chi_1 dihedral restraints: 126 0 2 73.628 73.628 28.504 1.000
15 Sidechain Chi_2 dihedral restraints: 93 0 2 82.135 82.135 36.254 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 80.911 80.911 25.645 1.000
17 Sidechain Chi_4 dihedral restraints: 21 0 0 90.433 90.433 17.164 1.000
18 Disulfide distance restraints : 4 0 0 0.007 0.007 0.30987E-01 1.000
19 Disulfide angle restraints : 8 0 0 2.784 2.784 1.3690 1.000
20 Disulfide dihedral angle restraints: 4 0 0 28.179 28.179 2.7141 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1582 0 0 0.414 0.414 48.181 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 144 20 21 24.562 65.031 72.156 1.000
26 Distance restraints 4 (SDCH-SDCH) : 744 0 1 0.739 0.739 63.776 1.000
27 Distance restraints 5 (X-Y) : 1401 0 0 0.033 0.033 13.273 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: BUFGA.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 16770.9082
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 8092 63P 87P N O 499 712 14.47 10.99 3.49 4.57 10.99 3.49 4.57
2 8128 67S 87P N O 530 712 8.62 6.49 2.13 4.69 6.49 2.13 4.69
-------------------------------------------------------------------------------------------------
Feature 13 : Mainchain Omega dihedral restraints
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4354 63P 63P CA C 500 504 -157.49 -180.00 22.51 4.50 -180.00 22.51 4.50
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 1S 2G C N 5 7 -76.15 -80.20 5.71 0.18 82.20 -133.77 7.00
1 2G 2G N CA 7 8 170.08 174.10 8.50
2 4149 2G 3K C N 9 11 -66.44 -70.20 5.65 0.34 -62.90 177.01 22.80
2 3K 3K N CA 11 12 136.18 140.40 -40.80
3 4150 3K 4Y C N 18 20 -126.12 -124.30 7.68 0.34 -63.50 -175.33 32.45
3 4Y 4Y N CA 20 21 142.87 135.40 -43.40
4 4152 5I 6S C N 38 40 -81.80 -72.40 26.05 1.25 -64.10 149.35 11.96
4 6S 6S N CA 40 41 176.70 152.40 -35.00
5 4153 6S 7W C N 44 46 -62.24 -71.30 10.38 0.62 -63.00 178.13 22.32
5 7W 7W N CA 46 47 133.93 139.00 -44.20
6 4154 7W 8E C N 58 60 -90.96 -117.80 49.66 1.83 -63.60 138.05 17.05
6 8E 8E N CA 60 61 95.01 136.80 -40.30
7 4155 8E 9D C N 67 69 -148.64 -96.50 53.64 2.20 -63.30 -172.67 19.88
7 9D 9D N CA 69 70 126.76 114.20 -40.00
8 4156 9D 10S C N 75 77 -71.24 -72.40 12.03 0.75 -64.10 160.78 12.09
8 10S 10S N CA 77 78 164.37 152.40 -35.00
9 4158 11C 12S C N 87 89 168.34 -136.60 62.30 1.91 -64.10 -167.15 23.15
9 12S 12S N CA 89 90 -179.64 151.20 -35.00
10 4159 12S 13Y C N 93 95 -122.71 -124.30 16.97 0.97 -63.50 174.64 30.69
10 13Y 13Y N CA 95 96 152.30 135.40 -43.40
11 4160 13Y 14L C N 105 107 -79.05 -108.50 57.75 2.74 -63.50 125.00 16.53
11 14L 14L N CA 107 108 82.83 132.50 -41.20
12 4161 14L 15Q C N 113 115 -169.29 -121.10 51.23 1.63 -63.80 -166.17 33.83
12 15Q 15Q N CA 115 116 157.09 139.70 -40.30
13 4162 15Q 16L C N 122 124 -110.32 -108.50 3.42 0.19 -63.50 177.11 22.38
13 16L 16L N CA 124 125 129.61 132.50 -41.20
14 4163 16L 17Q C N 130 132 -95.98 -73.00 48.67 3.63 -63.80 141.80 19.31
14 17Q 17Q N CA 132 133 97.80 140.70 -40.30
15 4198 51E 52S C N 414 416 -137.42 -64.10 82.36 8.50 -64.10 82.36 8.50
15 52S 52S N CA 416 417 2.50 -35.00 -35.00
16 4209 62N 63P C N 497 499 -51.02 -64.50 23.65 1.39 -58.70 158.45 13.72
16 63P 63P N CA 499 500 127.76 147.20 -30.50
17 4233 86T 87P C N 704 706 -43.25 -58.70 65.66 4.31 -64.50 120.38 10.13
17 87P 87P N CA 706 707 -94.31 -30.50 147.20
18 4234 87P 88R C N 711 713 -97.82 -125.20 61.09 2.25 57.30 162.37 16.28
18 88R 88R N CA 713 714 86.00 140.60 38.00
19 4288 141T 142Q C N 1128 1130 -49.51 -63.80 29.42 3.78 -73.00 155.07 10.62
19 142Q 142Q N CA 1130 1131 -66.02 -40.30 140.70
20 4289 142Q 143G C N 1137 1139 -65.40 -62.40 21.96 3.14 82.20 150.23 10.96
20 143G 143G N CA 1139 1140 -19.45 -41.20 8.50
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 10 15 53 84 141 146 149 171 193 213
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 149
Number of all, selected real atoms : 1209 1209
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 13409 13409
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2542
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 983.3759
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1197 0 0 0.006 0.006 13.093 1.000
2 Bond angle potential : 1618 5 12 3.614 3.614 237.92 1.000
3 Stereochemical cosine torsion poten: 814 0 33 47.531 47.531 285.24 1.000
4 Stereochemical improper torsion pot: 502 0 0 1.295 1.295 19.267 1.000
5 Soft-sphere overlap restraints : 2542 1 2 0.007 0.007 16.395 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2405 0 0 0.109 0.109 31.571 1.000
10 Distance restraints 2 (N-O) : 2564 1 7 0.207 0.207 106.09 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 145 0 1 4.355 4.355 32.427 1.000
14 Sidechain Chi_1 dihedral restraints: 126 0 1 68.218 68.218 22.555 1.000
15 Sidechain Chi_2 dihedral restraints: 93 0 1 72.801 72.801 37.824 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 75.372 75.372 20.889 1.000
17 Sidechain Chi_4 dihedral restraints: 21 0 0 72.085 72.085 13.159 1.000
18 Disulfide distance restraints : 4 0 0 0.008 0.008 0.46699E-01 1.000
19 Disulfide angle restraints : 8 0 0 1.945 1.945 0.66818 1.000
20 Disulfide dihedral angle restraints: 4 0 0 29.915 29.915 3.3518 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1582 0 0 0.364 0.364 29.176 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 144 19 16 24.152 62.686 58.989 1.000
26 Distance restraints 4 (SDCH-SDCH) : 744 0 1 0.640 0.640 43.014 1.000
27 Distance restraints 5 (X-Y) : 1401 0 0 0.030 0.030 11.707 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: BUFGA.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 15708.2383
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2136 82N 82N ND2 CG 667 665 138.90 122.50 16.40 4.57 122.50 16.40 4.57
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 8128 67S 87P N O 530 712 8.58 6.49 2.09 4.60 6.49 2.09 4.60
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 1S 2G C N 5 7 -87.38 -80.20 10.00 0.33 82.20 -118.06 7.50
1 2G 2G N CA 7 8 -178.94 174.10 8.50
2 4149 2G 3K C N 9 11 -76.75 -70.20 9.49 0.57 -62.90 172.49 23.18
2 3K 3K N CA 11 12 147.27 140.40 -40.80
3 4150 3K 4Y C N 18 20 -116.17 -124.30 8.16 0.59 -63.50 -174.35 26.98
3 4Y 4Y N CA 20 21 134.62 135.40 -43.40
4 4152 5I 6S C N 38 40 -65.80 -72.40 26.92 1.37 -64.10 161.32 11.59
4 6S 6S N CA 40 41 126.31 152.40 -35.00
5 4153 6S 7W C N 44 46 -150.33 -124.90 25.76 1.03 -63.00 -170.42 29.58
5 7W 7W N CA 46 47 147.53 143.40 -44.20
6 4154 7W 8E C N 58 60 -86.34 -69.30 28.75 2.63 -63.60 161.25 20.33
6 8E 8E N CA 60 61 119.34 142.50 -40.30
7 4155 8E 9D C N 67 69 -87.00 -96.50 19.16 0.81 -63.30 139.59 15.93
7 9D 9D N CA 69 70 97.57 114.20 -40.00
8 4156 9D 10S C N 75 77 179.68 -136.60 47.46 1.49 -64.10 -165.98 22.45
8 10S 10S N CA 77 78 169.64 151.20 -35.00
9 4158 11C 12S C N 87 89 -83.92 -72.40 86.85 5.51 -64.10 103.24 6.57
9 12S 12S N CA 89 90 66.32 152.40 -35.00
10 4159 12S 13Y C N 93 95 -143.54 -124.30 33.17 1.33 -63.50 173.71 31.45
10 13Y 13Y N CA 95 96 162.42 135.40 -43.40
11 4160 13Y 14L C N 105 107 -78.44 -70.70 25.29 2.22 -63.50 159.42 21.32
11 14L 14L N CA 107 108 117.52 141.60 -41.20
12 4161 14L 15Q C N 113 115 -102.19 -121.10 75.22 3.22 -63.80 113.86 15.04
12 15Q 15Q N CA 115 116 66.90 139.70 -40.30
13 4162 15Q 16L C N 122 124 -143.64 -108.50 35.70 1.63 -63.50 -162.98 32.32
13 16L 16L N CA 124 125 138.81 132.50 -41.20
14 4163 16L 17Q C N 130 132 -113.61 -121.10 12.43 0.43 -63.80 177.23 23.75
14 17Q 17Q N CA 132 133 129.78 139.70 -40.30
15 4198 51E 52S C N 414 416 -139.99 -64.10 85.39 8.79 -64.10 85.39 8.79
15 52S 52S N CA 416 417 4.14 -35.00 -35.00
16 4209 62N 63P C N 497 499 -67.46 -64.50 60.13 4.33 -58.70 122.56 10.85
16 63P 63P N CA 499 500 -152.74 147.20 -30.50
17 4210 63P 64P C N 504 506 -58.27 -58.70 26.11 2.19 -64.50 151.72 11.00
17 64P 64P N CA 506 507 -4.39 -30.50 147.20
18 4233 86T 87P C N 704 706 -42.92 -58.70 65.80 4.30 -64.50 120.37 10.15
18 87P 87P N CA 706 707 -94.38 -30.50 147.20
19 4234 87P 88R C N 711 713 -102.69 -125.20 57.12 2.19 57.30 167.66 16.77
19 88R 88R N CA 713 714 88.10 140.60 38.00
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 8 13 62 90 112 119 133 188 179 198
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 149
Number of all, selected real atoms : 1209 1209
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 13409 13409
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2550
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1074.4081
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1197 0 0 0.006 0.006 13.757 1.000
2 Bond angle potential : 1618 0 6 2.346 2.346 175.57 1.000
3 Stereochemical cosine torsion poten: 814 0 29 46.476 46.476 275.49 1.000
4 Stereochemical improper torsion pot: 502 0 0 1.314 1.314 20.117 1.000
5 Soft-sphere overlap restraints : 2550 1 2 0.007 0.007 15.617 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2405 0 0 0.132 0.132 45.703 1.000
10 Distance restraints 2 (N-O) : 2564 3 19 0.267 0.267 186.65 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 145 0 4 4.701 4.701 37.783 1.000
14 Sidechain Chi_1 dihedral restraints: 126 0 1 70.365 70.365 32.198 1.000
15 Sidechain Chi_2 dihedral restraints: 93 0 1 78.971 78.971 39.809 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 74.832 74.832 25.234 1.000
17 Sidechain Chi_4 dihedral restraints: 21 0 0 93.449 93.449 13.977 1.000
18 Disulfide distance restraints : 4 0 0 0.009 0.009 0.50262E-01 1.000
19 Disulfide angle restraints : 8 0 0 1.831 1.831 0.59202 1.000
20 Disulfide dihedral angle restraints: 4 0 0 29.606 29.606 3.3312 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1582 0 0 0.390 0.390 38.048 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 144 24 19 27.553 68.152 89.982 1.000
26 Distance restraints 4 (SDCH-SDCH) : 744 0 1 0.642 0.642 47.297 1.000
27 Distance restraints 5 (X-Y) : 1401 0 0 0.033 0.033 13.207 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: BUFGA.V99990003
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 17020.3633
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 7598 43N 36F N O 338 289 11.71 9.41 2.29 4.65 9.41 2.29 4.65
2 7627 44W 36F N O 346 289 11.08 8.62 2.46 5.45 8.62 2.46 5.45
3 8128 67S 87P N O 530 712 8.59 6.49 2.10 4.62 6.49 2.10 4.62
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 1S 2G C N 5 7 91.83 78.70 13.13 0.65 82.20 175.34 8.02
1 2G 2G N CA 7 8 -166.57 -166.10 8.50
2 4149 2G 3K C N 9 11 -65.80 -70.20 9.67 0.60 -62.90 172.61 22.26
2 3K 3K N CA 11 12 131.79 140.40 -40.80
3 4150 3K 4Y C N 18 20 -128.41 -124.30 15.58 0.67 -63.50 178.40 31.55
3 4Y 4Y N CA 20 21 150.43 135.40 -43.40
4 4152 5I 6S C N 38 40 -61.43 -72.40 11.06 0.83 -64.10 171.16 12.24
4 6S 6S N CA 40 41 153.86 152.40 -35.00
5 4153 6S 7W C N 44 46 -148.52 -124.90 106.63 4.59 -63.00 109.52 19.08
5 7W 7W N CA 46 47 -112.62 143.40 -44.20
6 4154 7W 8E C N 58 60 -67.98 -69.30 23.84 1.87 -63.60 153.46 20.70
6 8E 8E N CA 60 61 166.31 142.50 -40.30
7 4156 9D 10S C N 75 77 -77.32 -72.40 13.48 0.65 -64.10 160.59 12.47
7 10S 10S N CA 77 78 164.95 152.40 -35.00
8 4158 11C 12S C N 87 89 -173.92 -136.60 44.27 1.35 -64.10 -174.11 21.40
8 12S 12S N CA 89 90 175.02 151.20 -35.00
9 4159 12S 13Y C N 93 95 -79.07 -98.40 19.34 0.76 -63.50 171.69 26.46
9 13Y 13Y N CA 95 96 127.58 128.40 -43.40
10 4160 13Y 14L C N 105 107 -61.16 -70.70 19.23 1.19 -63.50 166.13 23.20
10 14L 14L N CA 107 108 124.91 141.60 -41.20
11 4161 14L 15Q C N 113 115 -125.23 -121.10 10.59 0.55 -63.80 -179.00 23.89
11 15Q 15Q N CA 115 116 129.95 139.70 -40.30
12 4162 15Q 16L C N 122 124 -124.03 -108.50 16.26 0.73 -63.50 -171.50 23.49
12 16L 16L N CA 124 125 137.32 132.50 -41.20
13 4163 16L 17Q C N 130 132 -105.68 -121.10 18.32 0.57 -63.80 175.19 23.77
13 17Q 17Q N CA 132 133 129.80 139.70 -40.30
14 4182 35D 36F C N 277 279 -61.04 -63.20 15.97 2.08 -124.20 168.83 9.13
14 36F 36F N CA 279 280 -60.13 -44.30 143.30
15 4183 36F 37K C N 288 290 -122.32 -118.00 82.22 4.05 56.60 179.87 20.22
15 37K 37K N CA 290 291 56.99 139.10 38.60
16 4198 51E 52S C N 414 416 -135.93 -64.10 79.19 8.37 -64.10 79.19 8.37
16 52S 52S N CA 416 417 -1.67 -35.00 -35.00
17 4209 62N 63P C N 497 499 -64.96 -64.50 59.59 4.42 -58.70 122.87 10.67
17 63P 63P N CA 499 500 -153.21 147.20 -30.50
18 4210 63P 64P C N 504 506 -57.82 -58.70 24.77 2.12 -64.50 153.09 11.08
18 64P 64P N CA 506 507 -5.74 -30.50 147.20
19 4233 86T 87P C N 704 706 -44.07 -58.70 63.52 4.19 -64.50 122.21 10.23
19 87P 87P N CA 706 707 -92.31 -30.50 147.20
20 4234 87P 88R C N 711 713 -95.19 -72.10 60.52 5.09 57.30 159.86 15.98
20 88R 88R N CA 713 714 85.96 141.90 38.00
21 4265 118D 119P C N 942 944 -65.88 -64.50 31.07 2.39 -58.70 146.83 11.56
21 119P 119P N CA 944 945 116.16 147.20 -30.50
22 4266 119P 120N C N 949 951 151.06 -63.20 145.74 22.07 -63.20 145.74 22.07
22 120N 120N N CA 951 952 -40.89 -41.10 -41.10
23 4279 132C 133K C N 1054 1056 -52.22 -62.90 29.19 3.38 -70.20 152.69 11.48
23 133K 133K N CA 1056 1057 -67.97 -40.80 140.40
24 4280 133K 134G C N 1063 1065 -73.90 -62.40 14.35 2.11 82.20 161.43 12.05
24 134G 134G N CA 1065 1066 -32.62 -41.20 8.50
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 8 13 51 84 118 133 144 185 172 208
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
BUFGA.B99990001.pdb 1045.65540
BUFGA.B99990002.pdb 983.37592
BUFGA.B99990003.pdb 1074.40808
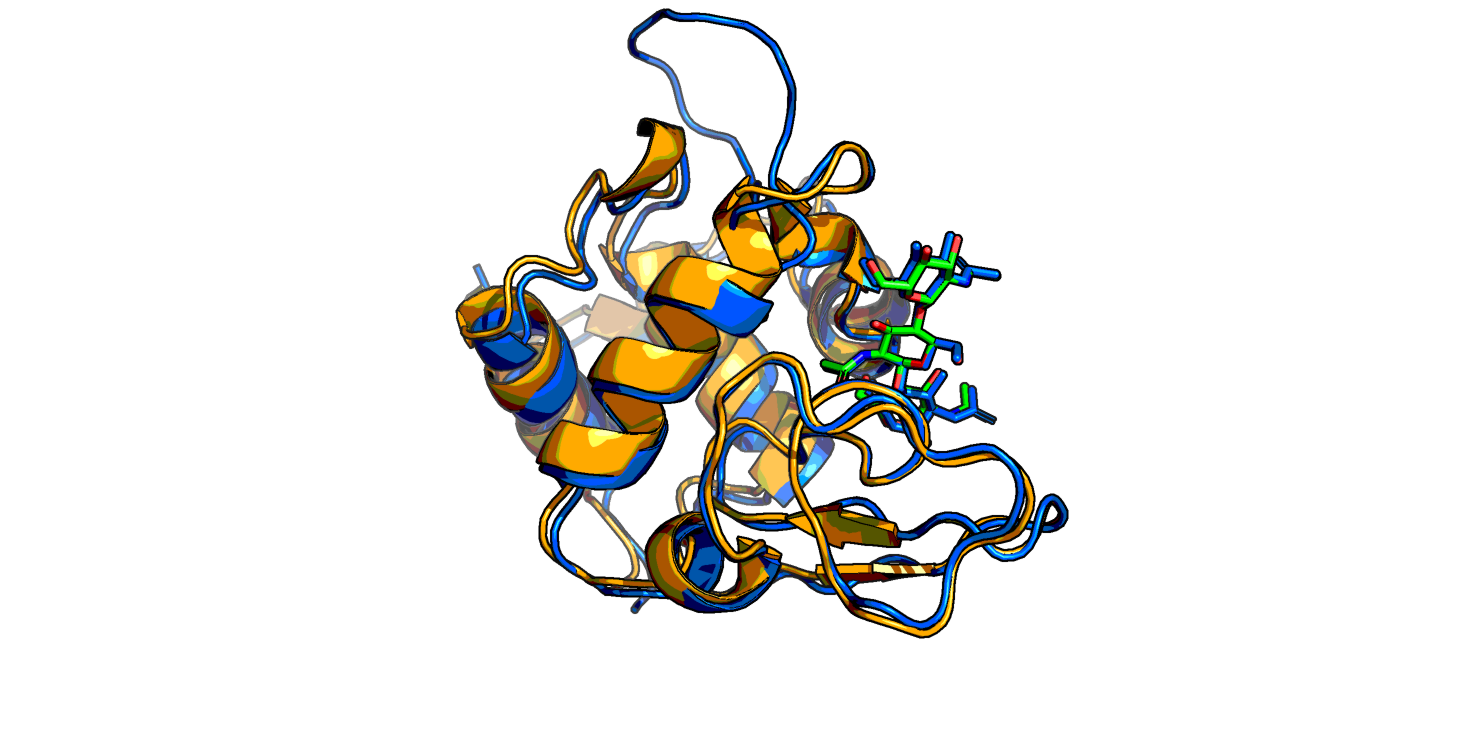
Получили 3 модели лизоцима жабы. Между собой модели практически не отличаются, хорошо ложатся на структуру лизоцима форели. Кое-где разметка вторичной структуры длиннее, чем у форели, а также присутствует неструктурированный "хвост" (Рис. 1). И еще у жабьего лизоцима присутствует маленький бета-тяж там, где у форели его нет.
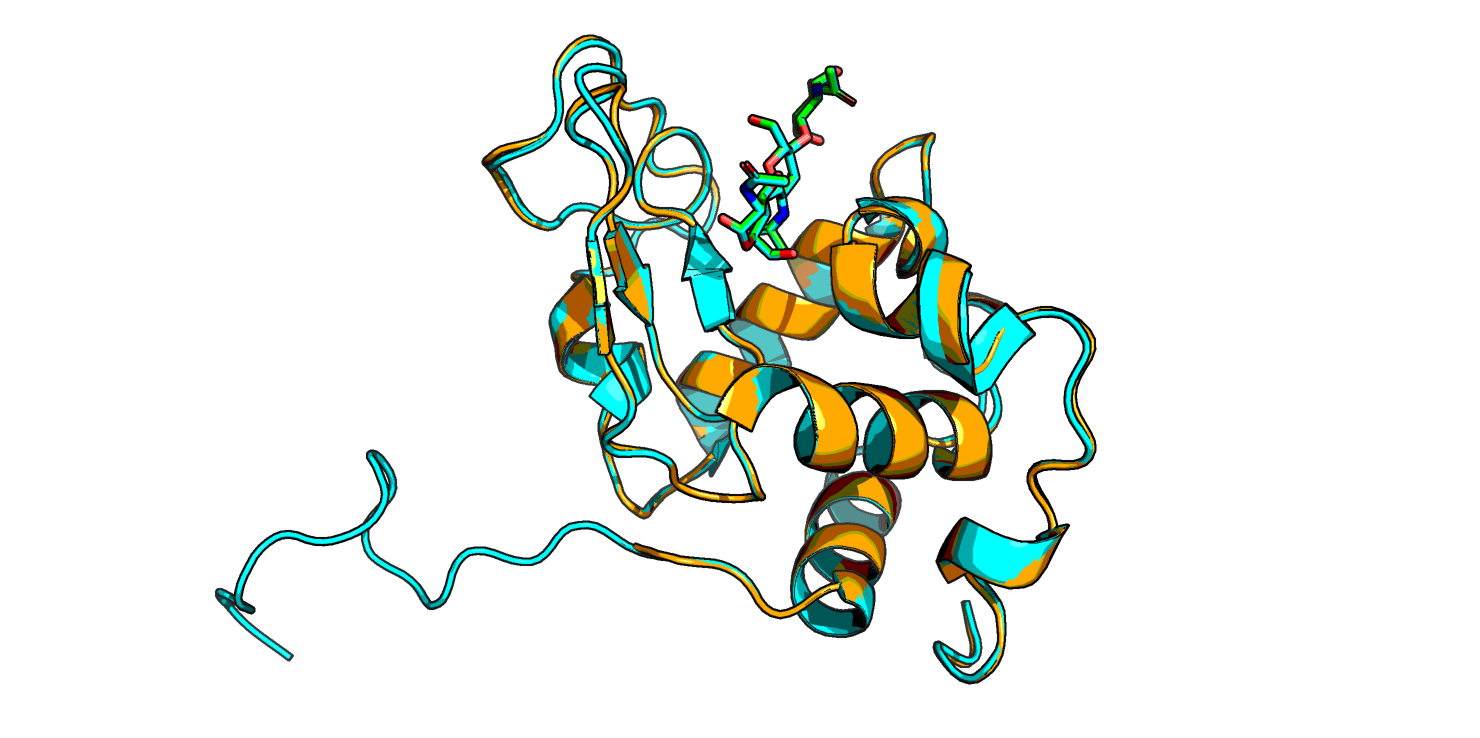
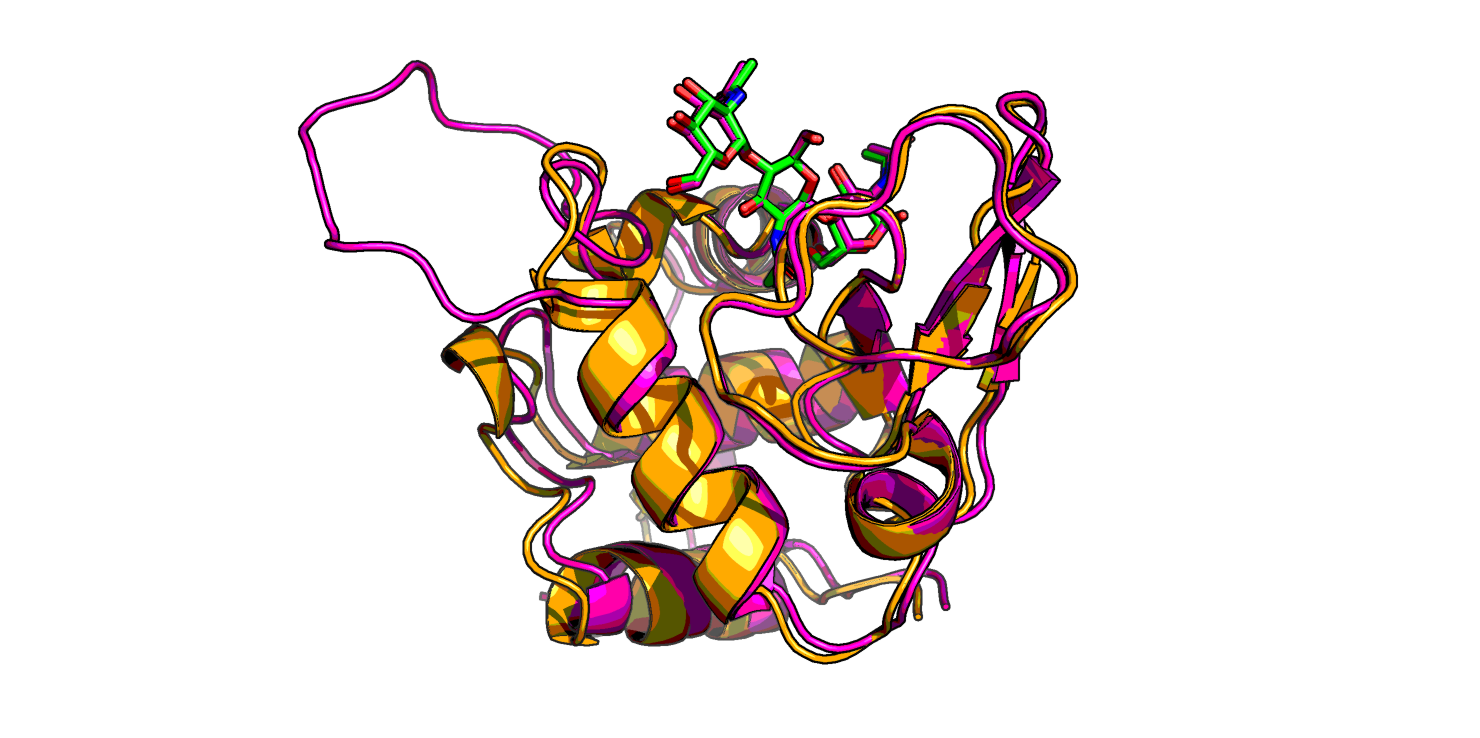
Теперь поместим лиганд в другое место белка:
! rm BUFGA.rsr
class mymodel(modeller.automodel.automodel):
def special_restraints(self, aln):
rsr = self.restraints
at = self.atoms
for x,y in [('O6:149', 'CA:140')]:
rsr.add(modeller.forms.gaussian(group=modeller.physical.xy_distance,
feature=modeller.features.distance(at[x],at[y]),
mean=3.0, stdev=0.1))
from modeller import *
from modeller.automodel import *
a = mymodel(env, alnfile='BUFGA_lig.ali', knowns= pdb.code, sequence = s.code)
a.make()
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 10 146
atom names : C +N
atom indices : 1164 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 10 146
atom names : C CA +N O
atom indices : 1164 1159 0 1165
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 4
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 14446 13410
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 149
Number of all, selected real atoms : 1209 1209
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 13410 13410
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 3076
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 7310.0532
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1197 4 12 0.019 0.019 120.26 1.000
2 Bond angle potential : 1618 11 37 3.928 3.928 422.34 1.000
3 Stereochemical cosine torsion poten: 814 0 34 48.130 48.130 293.75 1.000
4 Stereochemical improper torsion pot: 502 1 5 2.305 2.305 56.020 1.000
5 Soft-sphere overlap restraints : 3076 4 8 0.015 0.015 79.860 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2405 105 193 1.050 1.050 3427.9 1.000
10 Distance restraints 2 (N-O) : 2564 75 151 0.777 0.777 1983.8 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 145 1 8 5.879 5.879 59.107 1.000
14 Sidechain Chi_1 dihedral restraints: 126 0 3 78.216 78.216 49.353 1.000
15 Sidechain Chi_2 dihedral restraints: 93 0 0 65.481 65.481 36.664 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 67.575 67.575 20.402 1.000
17 Sidechain Chi_4 dihedral restraints: 21 0 0 90.106 90.106 14.887 1.000
18 Disulfide distance restraints : 4 0 0 0.008 0.008 0.43613E-01 1.000
19 Disulfide angle restraints : 8 0 0 3.582 3.582 2.2661 1.000
20 Disulfide dihedral angle restraints: 4 0 0 30.325 30.325 3.3593 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1582 0 0 0.498 0.498 70.774 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 144 20 19 27.008 65.082 84.311 1.000
26 Distance restraints 4 (SDCH-SDCH) : 744 1 6 0.937 0.937 105.85 1.000
27 Distance restraints 5 (X-Y) : 1402 1 1 0.122 0.122 479.04 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: BUFGA.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 25118.3867
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 1 : Bond length potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1122 137L 137L C CA 1092 1087 1.66 1.49 0.17 5.03 1.49 0.17 5.03
2 1135 139Q 139Q N CA 1100 1101 1.58 1.43 0.15 5.04 1.43 0.15 5.04
3 1137 139Q 139Q C CA 1107 1101 1.66 1.49 0.17 4.84 1.49 0.17 4.84
4 1150 140W 140W C CA 1121 1110 1.85 1.49 0.36 10.35 1.49 0.36 10.35
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1721 47T 47T CA C 375 379 137.95 116.50 21.45 6.17 116.50 21.45 6.17
2 1727 48A 48A N CA 381 382 126.28 107.00 19.28 5.54 107.00 19.28 5.54
3 2718 137L 137L N CA 1086 1087 124.69 107.00 17.69 5.09 107.00 17.69 5.09
4 2729 138S 138S N CA 1094 1095 124.88 107.00 17.88 5.14 107.00 17.88 5.14
5 2734 138S 139Q C N 1098 1100 148.74 120.00 28.74 6.53 120.00 28.74 6.53
6 2737 139Q 139Q N CA 1100 1101 130.08 107.00 23.08 6.64 107.00 23.08 6.64
7 2752 140W 140W CA C 1110 1121 134.31 116.50 17.81 5.12 116.50 17.81 5.12
-------------------------------------------------------------------------------------------------
Feature 9 : Distance restraints 1 (CA-CA)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4833 20E 140W CA CA 163 1110 14.48 11.22 3.26 5.58 11.22 3.26 5.58
2 4867 21R 140W CA CA 172 1110 11.57 7.44 4.13 10.25 7.44 4.13 10.25
3 4895 22C 139Q CA CA 183 1101 14.60 10.88 3.72 7.21 10.88 3.72 7.21
4 4896 22C 140W CA CA 183 1110 13.35 8.34 5.01 12.93 8.34 5.01 12.93
5 4897 22C 141T CA CA 183 1124 9.10 6.67 2.43 6.62 6.67 2.43 6.62
6 4926 23E 140W CA CA 189 1110 14.65 11.55 3.10 6.82 11.55 3.10 6.82
7 5007 25A 140W CA CA 206 1110 11.25 8.40 2.85 8.20 8.40 2.85 8.20
8 5035 26K 140W CA CA 211 1110 14.73 11.87 2.86 5.97 11.87 2.86 5.97
9 5162 30K 146L CA CA 242 1159 12.19 9.62 2.57 5.41 9.62 2.57 5.41
10 5261 34A 138S CA CA 267 1095 16.63 13.58 3.04 4.69 13.58 3.04 4.69
11 5399 40S 138S CA CA 316 1095 13.90 10.76 3.14 4.83 10.76 3.14 4.83
12 5424 41L 138S CA CA 322 1095 12.72 9.89 2.82 5.11 9.89 2.82 5.11
13 5426 41L 140W CA CA 322 1110 11.01 8.83 2.18 4.88 8.83 2.18 4.88
14 5453 42E 138S CA CA 330 1095 9.74 6.81 2.93 5.69 6.81 2.93 5.69
15 5454 42E 139Q CA CA 330 1101 10.98 8.17 2.81 5.41 8.17 2.81 5.41
16 5455 42E 140W CA CA 330 1110 10.27 5.67 4.60 10.58 5.67 4.60 10.58
17 5456 42E 141T CA CA 330 1124 8.58 6.10 2.48 5.79 6.10 2.48 5.79
18 7078 135K 140W CA CA 1070 1110 14.41 10.95 3.46 7.08 10.95 3.46 7.08
19 7081 136N 139Q CA CA 1079 1101 10.55 7.49 3.06 5.63 7.49 3.06 5.63
20 7082 136N 140W CA CA 1079 1110 13.24 9.04 4.20 8.17 9.04 4.20 8.17
21 7085 137L 139Q CA CA 1087 1101 7.66 5.37 2.29 5.92 5.37 2.29 5.92
22 7086 137L 140W CA CA 1087 1110 9.76 5.61 4.16 12.10 5.61 4.16 12.10
23 7087 137L 141T CA CA 1087 1124 9.63 7.77 1.86 5.23 7.77 1.86 5.23
24 7091 138S 140W CA CA 1095 1110 7.58 5.53 2.05 5.14 5.53 2.05 5.14
25 7092 138S 141T CA CA 1095 1124 8.57 6.03 2.54 6.23 6.03 2.54 6.23
26 7099 139Q 142Q CA CA 1101 1131 7.23 4.97 2.26 5.64 4.97 2.26 5.64
27 7106 140W 144C CA CA 1110 1144 11.50 8.83 2.67 7.34 8.83 2.67 7.34
28 7108 140W 146L CA CA 1110 1159 13.88 11.22 2.66 5.98 11.22 2.66 5.98
29 7110 141T 144C CA CA 1124 1144 8.01 5.55 2.46 7.46 5.55 2.46 7.46
30 7112 141T 146L CA CA 1124 1159 10.37 7.48 2.89 7.18 7.48 2.89 7.18
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 7195 21R 139Q N O 171 1108 12.08 9.22 2.86 4.99 9.22 2.86 4.99
2 7196 21R 140W N O 171 1122 10.49 7.02 3.47 7.92 7.02 3.47 7.92
3 7213 22C 139Q N O 182 1108 12.54 8.30 4.24 8.26 8.30 4.24 8.26
4 7214 22C 140W N O 182 1122 10.66 5.79 4.87 12.76 5.79 4.87 12.76
5 7229 23E 139Q N O 188 1108 14.13 10.92 3.22 5.45 10.92 3.22 5.45
6 7230 23E 140W N O 188 1122 11.65 8.09 3.56 8.08 8.09 3.56 8.08
7 7276 25A 140W N O 205 1122 8.92 6.63 2.29 6.58 6.63 2.29 6.58
8 7296 26K 140W N O 210 1122 11.35 8.36 2.99 6.43 8.36 2.99 6.43
9 7319 27A 144C N O 219 1148 10.79 8.90 1.89 4.50 8.90 1.89 4.50
10 7382 30K 144C N O 241 1148 13.48 10.60 2.89 5.85 10.60 2.89 5.85
11 7560 41L 137L N O 321 1093 10.94 8.51 2.43 5.57 8.51 2.43 5.57
12 7562 41L 140W N O 321 1122 11.73 9.57 2.16 4.67 9.57 2.16 4.67
13 7585 42E 137L N O 329 1093 8.69 5.74 2.95 6.41 5.74 2.95 6.41
14 7586 42E 138S N O 329 1099 10.57 7.72 2.85 4.85 7.72 2.85 4.85
15 7587 42E 139Q N O 329 1108 12.00 9.19 2.81 4.89 9.19 2.81 4.89
16 7588 42E 140W N O 329 1122 10.55 6.96 3.59 7.58 6.96 3.59 7.58
17 7589 42E 141T N O 329 1129 10.62 8.47 2.15 4.56 8.47 2.15 4.56
18 9557 137L 139Q N O 1086 1108 10.21 7.94 2.27 4.69 7.94 2.27 4.69
19 9558 137L 140W N O 1086 1122 11.98 9.10 2.88 7.19 9.10 2.88 7.19
20 9583 140W 21R N O 1109 181 11.51 8.54 2.97 6.39 8.54 2.97 6.39
21 9584 140W 22C N O 1109 187 14.40 10.91 3.48 7.62 10.91 3.48 7.62
22 9587 140W 42E N O 1109 337 8.38 6.17 2.21 4.70 6.17 2.21 4.70
23 9594 140W 135K N O 1109 1077 12.13 9.23 2.90 5.76 9.23 2.90 5.76
24 9595 140W 136N N O 1109 1085 9.99 5.93 4.06 7.93 5.93 4.06 7.93
25 9596 140W 137L N O 1109 1093 6.43 2.93 3.50 10.10 2.93 3.50 10.10
26 9602 141T 22C N O 1123 187 10.82 8.66 2.16 5.03 8.66 2.16 5.03
27 9613 141T 136N N O 1123 1085 11.08 8.28 2.81 5.12 8.28 2.81 5.12
28 9614 141T 137L N O 1123 1093 7.43 4.11 3.32 9.65 4.11 3.32 9.65
29 9615 141T 138S N O 1123 1099 5.94 3.24 2.70 6.44 3.24 2.70 6.44
30 9620 141T 146L N O 1123 1165 12.43 9.23 3.20 6.84 9.23 3.20 6.84
31 9654 144C 140W N O 1143 1122 9.15 6.80 2.35 6.54 6.80 2.35 6.54
32 9655 144C 141T N O 1143 1129 6.35 3.04 3.30 10.72 3.04 3.30 10.72
33 9667 145K 141T N O 1149 1129 8.00 4.57 3.43 6.64 4.57 3.43 6.64
34 9678 146L 140W N O 1158 1122 11.57 9.35 2.22 4.86 9.35 2.22 4.86
35 9679 146L 141T N O 1158 1129 9.57 5.53 4.04 10.02 5.53 4.04 10.02
-------------------------------------------------------------------------------------------------
Feature 13 : Mainchain Omega dihedral restraints
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4427 136N 136N CA C 1079 1084 -156.30 -180.00 23.70 4.74 -180.00 23.70 4.74
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 1S 2G C N 5 7 -147.22 -167.20 20.02 0.41 82.20 -147.76 15.81
1 2G 2G N CA 7 8 175.82 174.60 8.50
2 4149 2G 3K C N 9 11 -71.50 -70.20 14.41 1.01 -62.90 164.68 21.87
2 3K 3K N CA 11 12 154.75 140.40 -40.80
3 4150 3K 4Y C N 18 20 -123.02 -124.30 18.89 1.05 -63.50 172.92 30.43
3 4Y 4Y N CA 20 21 154.25 135.40 -43.40
4 4152 5I 6S C N 38 40 -143.70 -136.60 19.99 0.84 -64.10 174.34 18.34
4 6S 6S N CA 40 41 169.88 151.20 -35.00
5 4153 6S 7W C N 44 46 -90.10 -71.30 36.93 3.16 -63.00 153.82 17.81
5 7W 7W N CA 46 47 107.22 139.00 -44.20
6 4154 7W 8E C N 58 60 -136.10 -117.80 18.44 0.73 -63.60 -170.69 22.19
6 8E 8E N CA 60 61 134.57 136.80 -40.30
7 4155 8E 9D C N 67 69 -104.77 -96.50 11.44 0.47 -63.30 152.07 16.70
7 9D 9D N CA 69 70 106.30 114.20 -40.00
8 4156 9D 10S C N 75 77 -103.26 -72.40 56.30 4.30 -64.10 145.68 9.02
8 10S 10S N CA 77 78 105.31 152.40 -35.00
9 4158 11C 12S C N 87 89 -96.00 -72.40 49.52 3.69 -64.10 147.36 9.27
9 12S 12S N CA 89 90 108.87 152.40 -35.00
10 4159 12S 13Y C N 93 95 -118.38 -124.30 18.81 0.78 -63.50 170.05 24.46
10 13Y 13Y N CA 95 96 117.55 135.40 -43.40
11 4160 13Y 14L C N 105 107 -122.35 -108.50 16.76 0.74 -63.50 -173.60 29.52
11 14L 14L N CA 107 108 141.94 132.50 -41.20
12 4161 14L 15Q C N 113 115 -103.00 -121.10 23.67 0.75 -63.80 169.35 23.03
12 15Q 15Q N CA 115 116 124.45 139.70 -40.30
13 4162 15Q 16L C N 122 124 -125.98 -108.50 17.56 0.82 -63.50 -173.88 23.13
13 16L 16L N CA 124 125 134.12 132.50 -41.20
14 4163 16L 17Q C N 130 132 -137.37 -121.10 16.51 0.68 -63.80 -168.12 25.08
14 17Q 17Q N CA 132 133 136.91 139.70 -40.30
15 4209 62N 63P C N 497 499 -61.97 -64.50 58.51 4.50 -58.70 123.89 10.50
15 63P 63P N CA 499 500 -154.35 147.20 -30.50
16 4210 63P 64P C N 504 506 -59.08 -58.70 26.52 2.16 -64.50 151.27 11.00
16 64P 64P N CA 506 507 -3.98 -30.50 147.20
17 4284 137L 138S C N 1092 1094 83.89 -136.60 157.81 4.84 -64.10 178.63 16.90
17 138S 138S N CA 1094 1095 -135.05 151.20 -35.00
18 4285 138S 139Q C N 1098 1100 -40.80 -63.80 50.49 6.52 -63.80 50.49 6.52
18 139Q 139Q N CA 1100 1101 -85.25 -40.30 -40.30
19 4288 141T 142Q C N 1128 1130 167.69 -121.10 90.96 2.85 -73.00 131.63 8.26
19 142Q 142Q N CA 1130 1131 -163.70 139.70 140.70
20 4289 142Q 143G C N 1137 1139 -62.38 -62.40 15.33 2.29 82.20 148.61 11.01
20 143G 143G N CA 1139 1140 -25.87 -41.20 8.50
-------------------------------------------------------------------------------------------------
Feature 26 : Distance restraints 4 (SDCH-SDCH)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 11453 42E 140W CB CB 331 1111 11.44 4.63 6.80 4.78 4.63 6.80 4.78
-------------------------------------------------------------------------------------------------
Feature 27 : Distance restraints 5 (X-Y)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 13410 149. 140W O6 CA 1206 1110 6.78 3.00 3.78 37.78 3.00 3.78 37.78
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 2 6 14 47 92 109 158 163 171 198 220 244
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
BUFGA.B99990001.pdb 7310.05322
Скор у новой модели сильно выше, чем у трех первых: 7310 против ~1000. На Рис. 2 видно, что в новой модели пропал бета-тяж, которого не было в лизоциме форели, а еще по-другому структурирован C-конец (в левой нижней части изображения) - отсутствует маленькая спираль, и неструктурированные хвосты иначе расположены.
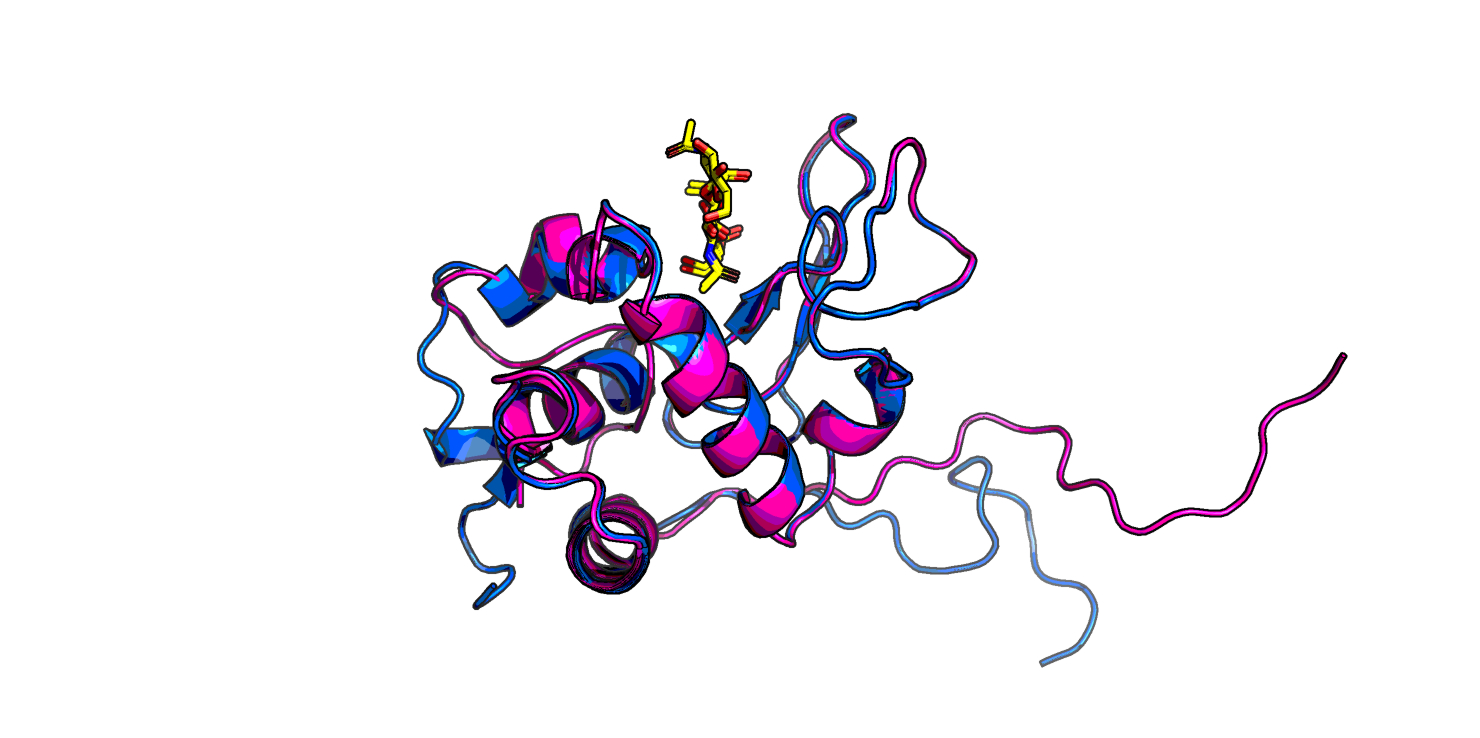
Теперь сделаем полиаланиновый лизоцим.
with open("P85045.fasta", 'r') as filik:
lines = filik.read().split("1\n") ##lines[0] is the string with identificator
prot = lines[1].replace("\n", "") ##join the lines of protein sequence into one
Ala = 'A'*len(prot)
with open("polyala.fasta", 'w') as polyala: ##make a new file with old identificator and polyalanine peptide
polyala.write(lines[0] + "1\n" + Ala + "\n")
! cat polyala.fasta
>sp|P85045|LYS_BUFGA Lysozyme C (Fragment) OS=Bufo gargarizans andrewsi OX=61428 PE=1 SV=1 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
alignm=modeller.alignment(env)
alignm.append(file='polyala.fasta', align_codes='all',alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
alignm[0].code = 'Lys_ALA'
alignm[1].code = '1LMP'
alignm.salign()
alignm.write(file='align_ala.ali', alignment_format='PIR')
SALIGN_____> adding the next group to the alignment; iteration 1
! cat align_ala.ali
>P1;Lys_ALA sequence:: : : : :::-1.00:-1.00 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGSTDYGIFQINSRYWCDDGRTPGAKNV CGIRCSQLLTDDLTVAIRCAKRV--------------VLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV...*
Снова надо добавить лиганд
with open("align_ala.ali", 'r') as align:
seq = align.read().split("*")
seq[0] = seq[0][:-2] + "..."
seq[1] = seq[1][:-3] + "-..."
new = "*".join(seq)
with open("align_ala_lig.ali", 'w') as out:
out.write(new)
! cat align_ala_lig.ali
>P1;Lys_ALA sequence:: : : : :::-1.00:-1.00 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA...* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGSTDYGIFQINSRYWCDDGRTPGAKNV CGIRCSQLLTDDLTVAIRCAKRV--------------VLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV-...*
Моделируем полиаланиновый белок на основе лизоцима форели.
## Выбираем объект для моделирования
s = alignm[0]
pdb = alignm[1]
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='align_ala_lig.ali', knowns= pdb.code , sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 2
a.make()
Lys_ALA 1LMP
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 1 144
atom names : C +N
atom indices : 719 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 1 144
atom names : C CA +N O
atom indices : 719 717 0 720
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 10455 9742
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 147
Number of all, selected real atoms : 764 764
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 9742 9742
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 1457
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 994.2429
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 720 0 0 0.009 0.009 16.795 1.000
2 Bond angle potential : 1007 2 16 2.786 2.786 158.17 1.000
3 Stereochemical cosine torsion poten: 292 0 37 77.067 77.067 285.11 1.000
4 Stereochemical improper torsion pot: 288 0 0 1.227 1.227 10.227 1.000
5 Soft-sphere overlap restraints : 1457 1 3 0.012 0.012 26.521 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2405 0 2 0.320 0.320 88.126 1.000
10 Distance restraints 2 (N-O) : 2564 1 15 0.485 0.485 179.27 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 143 0 4 5.095 5.095 43.768 1.000
14 Sidechain Chi_1 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
15 Sidechain Chi_2 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
16 Sidechain Chi_3 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
17 Sidechain Chi_4 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
18 Disulfide distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
19 Disulfide angle restraints : 0 0 0 0.000 0.000 0.0000 1.000
20 Disulfide dihedral angle restraints: 0 0 0 0.000 0.000 0.0000 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 718 0 0 0.254 0.254 5.3508 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 142 33 24 35.231 83.036 141.80 1.000
26 Distance restraints 4 (SDCH-SDCH) : 62 0 0 0.454 0.454 1.4309 1.000
27 Distance restraints 5 (X-Y) : 1401 0 3 0.058 0.058 37.674 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: Lys_ALA.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 15487.1865
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1016 43A 43A N CA 211 212 123.98 107.00 16.98 4.88 107.00 16.98 4.88
2 1509 113A 113A C CA 564 562 129.49 108.00 21.48 4.98 108.00 21.48 4.98
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 7071 113A 75A N O 561 375 15.11 10.72 4.39 4.51 10.72 4.39 4.51
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2322 15A 16A C N 74 76 79.06 55.40 27.06 2.04 -62.50 156.17 31.73
1 16A 16A N CA 76 77 25.06 38.20 -40.90
2 2325 18A 19A C N 89 91 61.90 55.40 16.83 0.61 -62.50 139.70 28.41
2 19A 19A N CA 91 92 22.68 38.20 -40.90
3 2327 20A 21A C N 99 101 61.51 -68.20 142.14 13.64 -62.50 169.57 26.42
3 21A 21A N CA 101 102 -156.56 145.30 -40.90
4 2343 36A 37A C N 179 181 65.31 55.40 12.15 0.81 -62.50 146.73 29.84
4 37A 37A N CA 181 182 31.18 38.20 -40.90
5 2344 37A 38A C N 184 186 60.04 55.40 8.71 0.34 -62.50 141.99 28.87
5 38A 38A N CA 186 187 30.83 38.20 -40.90
6 2349 42A 43A C N 209 211 -80.05 -62.50 17.56 3.35 -134.00 -178.98 11.09
6 43A 43A N CA 211 212 -40.20 -40.90 147.00
7 2355 48A 49A C N 239 241 85.89 55.40 42.14 2.30 -62.50 156.59 31.61
7 49A 49A N CA 241 242 9.12 38.20 -40.90
8 2360 53A 54A C N 264 266 91.74 -134.00 139.64 3.40 -62.50 -155.86 32.08
8 54A 54A N CA 266 267 -174.63 147.00 -40.90
9 2363 56A 57A C N 279 281 60.54 55.40 12.10 1.14 -62.50 152.47 30.89
9 57A 57A N CA 281 282 49.15 38.20 -40.90
10 2373 66A 67A C N 329 331 82.74 55.40 60.43 2.19 -62.50 147.41 29.24
10 67A 67A N CA 331 332 -15.70 38.20 -40.90
11 2377 70A 71A C N 349 351 53.96 55.40 11.63 0.59 -62.50 147.57 29.84
11 71A 71A N CA 351 352 49.74 38.20 -40.90
12 2380 73A 74A C N 364 366 54.36 55.40 20.09 1.13 -62.50 153.26 30.89
12 74A 74A N CA 366 367 58.27 38.20 -40.90
13 2383 76A 77A C N 379 381 64.13 55.40 9.68 1.15 -62.50 151.56 30.78
13 77A 77A N CA 381 382 42.37 38.20 -40.90
14 2405 98A 99A C N 489 491 -41.05 -68.20 29.16 2.09 -62.50 176.87 30.08
14 99A 99A N CA 491 492 134.67 145.30 -40.90
15 2407 100A 101A C N 499 501 19.21 -68.20 91.60 8.57 -62.50 167.67 25.12
15 101A 101A N CA 501 502 172.68 145.30 -40.90
16 2408 101A 102A C N 504 506 -116.92 -134.00 18.16 0.42 -62.50 -173.61 33.44
16 102A 102A N CA 506 507 140.83 147.00 -40.90
17 2409 102A 103A C N 509 511 -79.77 -68.20 24.50 2.33 -62.50 165.51 26.33
17 103A 103A N CA 511 512 123.71 145.30 -40.90
18 2410 103A 104A C N 514 516 154.67 -134.00 71.85 2.07 -62.50 -142.86 42.97
18 104A 104A N CA 516 517 155.56 147.00 -40.90
19 2411 104A 105A C N 519 521 -128.69 -134.00 16.68 0.79 -62.50 -175.62 27.84
19 105A 105A N CA 521 522 131.19 147.00 -40.90
20 2412 105A 106A C N 524 526 172.62 -134.00 57.17 1.32 -62.50 -163.57 38.69
20 106A 106A N CA 526 527 167.48 147.00 -40.90
21 2413 106A 107A C N 529 531 -110.92 -134.00 29.80 0.80 -62.50 175.84 26.89
21 107A 107A N CA 531 532 128.14 147.00 -40.90
22 2414 107A 108A C N 534 536 176.62 -134.00 52.41 1.22 -62.50 -163.79 38.48
22 108A 108A N CA 536 537 164.56 147.00 -40.90
23 2415 108A 109A C N 539 541 -68.89 -68.20 10.90 0.90 -62.50 175.44 28.44
23 109A 109A N CA 541 542 134.42 145.30 -40.90
24 2416 109A 110A C N 544 546 -170.30 -134.00 41.47 0.96 -62.50 -173.60 36.23
24 110A 110A N CA 546 547 167.04 147.00 -40.90
25 2417 110A 111A C N 549 551 -125.42 -134.00 18.76 0.79 -62.50 -177.60 27.59
25 111A 111A N CA 551 552 130.31 147.00 -40.90
26 2418 111A 112A C N 554 556 -159.00 -134.00 37.41 2.22 -62.50 -173.10 28.02
26 112A 112A N CA 556 557 119.17 147.00 -40.90
27 2419 112A 113A C N 559 561 -67.28 -62.50 67.99 11.38 -62.50 67.99 11.38
27 113A 113A N CA 561 562 -108.72 -40.90 -40.90
28 2420 113A 114A C N 564 566 -111.03 -62.50 48.58 9.43 -62.50 48.58 9.43
28 114A 114A N CA 566 567 -43.07 -40.90 -40.90
29 2424 117A 118A C N 584 586 109.77 -134.00 129.90 2.96 -62.50 -153.37 33.64
29 118A 118A N CA 586 587 -155.00 147.00 -40.90
30 2437 130A 131A C N 649 651 66.00 -68.20 150.51 14.56 -62.50 166.35 26.31
30 131A 131A N CA 651 652 -146.54 145.30 -40.90
31 2438 131A 132A C N 654 656 70.05 -68.20 138.54 12.04 -68.20 138.54 12.04
31 132A 132A N CA 656 657 154.29 145.30 145.30
32 2446 139A 140A C N 694 696 68.79 55.40 36.25 1.33 -62.50 138.92 28.07
32 140A 140A N CA 696 697 4.51 38.20 -40.90
33 2448 141A 142A C N 704 706 61.80 55.40 11.00 0.46 -62.50 142.73 29.03
33 142A 142A N CA 706 707 29.25 38.20 -40.90
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 15 9 53 58 82 84 76 111 107 106
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 147
Number of all, selected real atoms : 764 764
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 9742 9742
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 1406
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1310.8405
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 720 0 2 0.010 0.010 19.991 1.000
2 Bond angle potential : 1007 2 14 2.798 2.798 164.13 1.000
3 Stereochemical cosine torsion poten: 292 0 34 75.643 75.643 275.07 1.000
4 Stereochemical improper torsion pot: 288 0 0 1.264 1.264 11.340 1.000
5 Soft-sphere overlap restraints : 1406 2 5 0.016 0.016 40.248 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2405 2 26 0.578 0.578 195.06 1.000
10 Distance restraints 2 (N-O) : 2564 6 30 0.688 0.688 262.15 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 143 1 8 5.798 5.798 56.685 1.000
14 Sidechain Chi_1 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
15 Sidechain Chi_2 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
16 Sidechain Chi_3 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
17 Sidechain Chi_4 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
18 Disulfide distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
19 Disulfide angle restraints : 0 0 0 0.000 0.000 0.0000 1.000
20 Disulfide dihedral angle restraints: 0 0 0 0.000 0.000 0.0000 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 718 0 0 0.261 0.261 5.4773 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 142 32 20 26.975 77.435 114.14 1.000
26 Distance restraints 4 (SDCH-SDCH) : 62 0 1 0.849 0.849 5.0941 1.000
27 Distance restraints 5 (X-Y) : 1401 5 8 0.124 0.124 161.45 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: Lys_ALA.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 15533.8623
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1506 113A 113A N CA 561 562 137.12 107.00 30.12 8.66 107.00 30.12 8.66
2 1548 119A 119A N CA 591 592 125.19 107.00 18.19 5.23 107.00 18.19 5.23
-------------------------------------------------------------------------------------------------
Feature 9 : Distance restraints 1 (CA-CA)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4583 76A 113A CA CA 377 562 16.02 11.67 4.35 5.22 11.67 4.35 5.22
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 7071 113A 75A N O 561 375 17.25 10.72 6.52 6.70 10.72 6.52 6.70
2 7072 113A 91A N O 561 455 15.16 8.93 6.23 5.48 8.93 6.23 5.48
3 7073 113A 92A N O 561 460 14.22 7.86 6.36 5.89 7.86 6.36 5.89
4 7075 113A 94A N O 561 470 12.26 5.21 7.06 4.54 5.21 7.06 4.54
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2322 15A 16A C N 74 76 78.18 55.40 26.62 1.93 -62.50 155.11 31.51
1 16A 16A N CA 76 77 24.44 38.20 -40.90
2 2325 18A 19A C N 89 91 62.51 55.40 13.27 0.51 -62.50 142.25 28.93
2 19A 19A N CA 91 92 27.00 38.20 -40.90
3 2343 36A 37A C N 179 181 66.85 55.40 14.16 0.93 -62.50 147.44 29.99
3 37A 37A N CA 181 182 29.87 38.20 -40.90
4 2344 37A 38A C N 184 186 62.28 55.40 11.20 0.49 -62.50 143.20 29.12
4 38A 38A N CA 186 187 29.36 38.20 -40.90
5 2355 48A 49A C N 239 241 86.08 55.40 41.87 2.33 -62.50 156.97 31.70
5 49A 49A N CA 241 242 9.71 38.20 -40.90
6 2360 53A 54A C N 264 266 90.59 -134.00 140.91 3.43 -62.50 -157.11 31.87
6 54A 54A N CA 266 267 -174.05 147.00 -40.90
7 2363 56A 57A C N 279 281 60.32 55.40 13.61 1.22 -62.50 153.33 31.05
7 57A 57A N CA 281 282 50.89 38.20 -40.90
8 2373 66A 67A C N 329 331 82.15 55.40 59.15 2.14 -62.50 147.02 29.20
8 67A 67A N CA 331 332 -14.56 38.20 -40.90
9 2377 70A 71A C N 349 351 51.34 55.40 16.25 0.68 -62.50 148.17 29.89
9 71A 71A N CA 351 352 53.94 38.20 -40.90
10 2380 73A 74A C N 364 366 54.83 55.40 18.75 1.08 -62.50 152.77 30.81
10 74A 74A N CA 366 367 56.94 38.20 -40.90
11 2383 76A 77A C N 379 381 64.89 55.40 10.27 1.22 -62.50 152.05 30.89
11 77A 77A N CA 381 382 42.11 38.20 -40.90
12 2405 98A 99A C N 489 491 -100.63 -68.20 38.01 3.72 -62.50 170.68 26.37
12 99A 99A N CA 491 492 125.47 145.30 -40.90
13 2406 99A 100A C N 494 496 -87.88 -68.20 19.86 1.59 -62.50 173.04 29.67
13 100A 100A N CA 496 497 147.94 145.30 -40.90
14 2407 100A 101A C N 499 501 -69.32 -68.20 5.18 0.46 -62.50 178.99 29.67
14 101A 101A N CA 501 502 140.25 145.30 -40.90
15 2408 101A 102A C N 504 506 -152.30 -134.00 19.48 0.45 -62.50 -171.79 35.64
15 102A 102A N CA 506 507 153.69 147.00 -40.90
16 2409 102A 103A C N 509 511 -70.06 -68.20 1.87 0.17 -62.50 174.21 28.93
16 103A 103A N CA 511 512 145.05 145.30 -40.90
17 2410 103A 104A C N 514 516 -72.82 -68.20 5.26 0.36 -62.50 171.60 28.64
17 104A 104A N CA 516 517 147.81 145.30 -40.90
18 2411 104A 105A C N 519 521 -151.87 -134.00 26.33 0.83 -62.50 176.99 33.77
18 105A 105A N CA 521 522 166.33 147.00 -40.90
19 2412 105A 106A C N 524 526 -80.72 -68.20 32.20 3.00 -62.50 157.59 24.99
19 106A 106A N CA 526 527 115.64 145.30 -40.90
20 2413 106A 107A C N 529 531 -133.18 -134.00 14.93 0.87 -62.50 172.35 32.03
20 107A 107A N CA 531 532 161.91 147.00 -40.90
21 2414 107A 108A C N 534 536 -59.50 -68.20 11.77 0.75 -62.50 178.30 29.37
21 108A 108A N CA 536 537 137.38 145.30 -40.90
22 2415 108A 109A C N 539 541 -78.63 -68.20 11.57 0.80 -62.50 169.55 28.61
22 109A 109A N CA 541 542 150.32 145.30 -40.90
23 2416 109A 110A C N 544 546 176.61 -134.00 63.14 1.67 -62.50 179.56 35.64
23 110A 110A N CA 546 547 -173.67 147.00 -40.90
24 2417 110A 111A C N 549 551 -61.21 -68.20 7.20 0.55 -62.50 175.51 28.70
24 111A 111A N CA 551 552 143.60 145.30 -40.90
25 2418 111A 112A C N 554 556 -112.06 -134.00 38.62 1.43 -62.50 163.79 24.93
25 112A 112A N CA 556 557 115.21 147.00 -40.90
26 2419 112A 113A C N 559 561 -84.35 -62.50 48.83 9.17 -62.50 48.83 9.17
26 113A 113A N CA 561 562 -84.57 -40.90 -40.90
27 2420 113A 114A C N 564 566 -117.60 -62.50 55.23 10.77 -62.50 55.23 10.77
27 114A 114A N CA 566 567 -44.74 -40.90 -40.90
28 2421 114A 115A C N 569 571 -117.12 -62.50 90.45 17.69 -68.20 112.85 7.48
28 115A 115A N CA 571 572 -113.00 -40.90 145.30
29 2424 117A 118A C N 584 586 112.98 55.40 148.52 5.41 -62.50 -175.24 32.64
29 118A 118A N CA 586 587 -98.70 38.20 -40.90
30 2437 130A 131A C N 649 651 49.38 55.40 11.63 0.44 -62.50 142.99 28.89
30 131A 131A N CA 651 652 48.14 38.20 -40.90
31 2446 139A 140A C N 694 696 69.18 55.40 35.98 1.31 -62.50 139.44 28.18
31 140A 140A N CA 696 697 4.97 38.20 -40.90
32 2448 141A 142A C N 704 706 57.30 55.40 2.30 0.16 -62.50 142.84 29.02
32 142A 142A N CA 706 707 36.90 38.20 -40.90
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 6 14 56 60 74 71 94 124 114 89
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
Lys_ALA.B99990001.pdb 994.24292
Lys_ALA.B99990002.pdb 1310.84045
У обеих моделей скор близок к скору, полученному для нативной последовательности лизоцима жабы (в районе 1000). У второй модели он даже выше: 1310, а у лучшей модели нативной последовательности 1074. Однако между собой 2 модели различаются: у первой модели (голубая на Рис. 3) отсутствуют бета-тяжи и немного по-другому расположена петля в середине структуры. У второй модели (розовая на Рис. 3), напротив, бета-тяжи длиннее, чем в лизоциме форели, присутствует еще один маленький бета-тяжик, а также отсутствует фрагмент альфа-спирали. Но помимо этого можно сказать, что в целом MODELLER сложил из полиаланиновой цепи структуру, очень похожую на лизоцим, что, вероятно, объясняется "универсальностью" аланина.