MAIN
TERMS
PROJECTS
ABOUT
Preparation of reads
Command table
| Command name | Command function |
| fastqc chr10.1.fastq | read quality control |
Read quality analysis
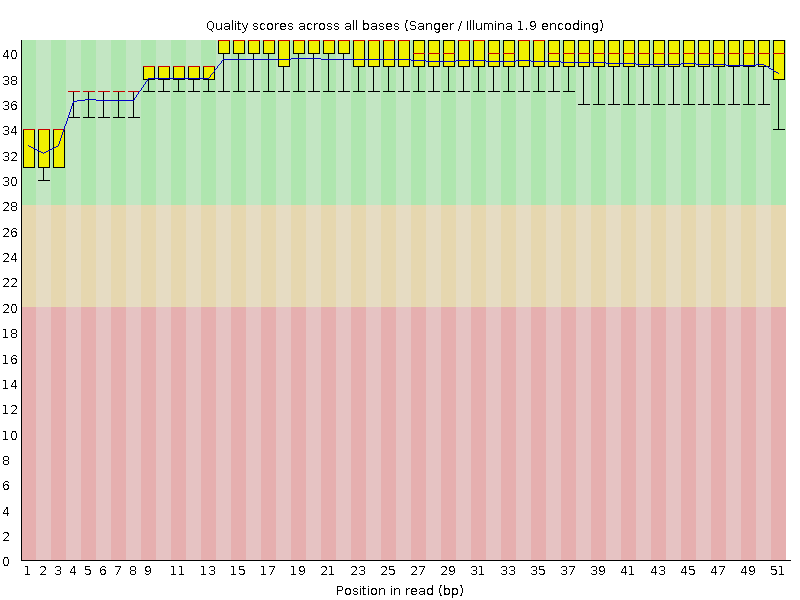
Per base quality chart before trimming
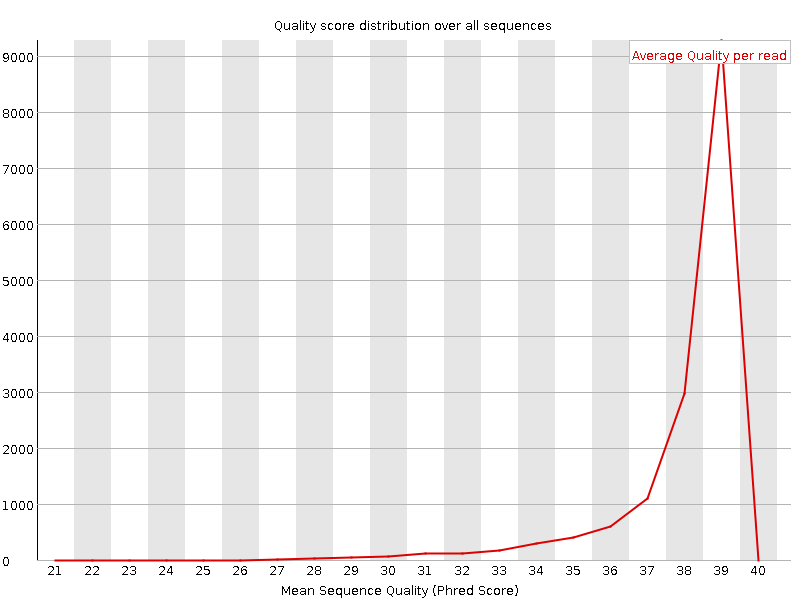
Per sequence quality scores
Number of reads: 15462
Mapping
| Command name | Command function |
| /home/students/y06/anastaisha_w/hisat2-2.0.5/hisat2-build chr10.fasta chr10 | reference indexing |
| /home/students/y06/anastaisha_w/hisat2-2.0.5/hisat2 -x chr10 -U chr10.1_out.fastq --no-softclip > chr10.1.sam | creating the alignment in .sam extension, no soft-clipped. chr10 - index file, chr10.1_out.fastq - input fastqc file |
| samtools view chr10.1.sam -b > chr10.1.bam | converting the alignment to .bam extension (binary file) |
| samtools sort chr10.1.bam -T temp.txt -o chr10.1_sorted.bam | sotring the .bam file, temp.txt - temporary file, chr10.1_sorted.bam - binary file |
| samtools index chr10.1_sorted.bam | indexing sorted .bam file |
Alignment result
15462 reads; of these:
15462 (100.00%) were unpaired; of these:
206 (1.33%) aligned 0 times
15194 (98.27%) aligned exactly 1 time
62 (0.40%) aligned >1 times
98.67% overall alignment rate
Here we can see, that 206 reads didn't map on the chromosome, 15257 reads were map.
Counting reads
| htseq-count option | htseq-count function |
| htseq-count -f | specifying the input format |
| htseq-count -s | specifying, whether the data is from a strand-specific assay. |
| htseq-count -i | specifying the attributee to be used as a feature ID |
| htseq-count -m | specifying the htseq-count mode |
| htseq-count -f bam -s no chr10.1_sorted.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf > final.txt | getting the htseq result |
After using the htseq-count with defaults and -f bam, we can see, that only one gene is presented in the output, meaning that all of the reads are placed to that gene.
braaist@kodomo:/nfs/srv/databases/ngs/braaist$ grep -wv 0 final.txt ENSG00000165732.8 14643 ENSG00000266122.1 3 __no_feature 610 __not_aligned 206
That was a gene of DExD-box helicase 21 protein with AC ENSG00000165732.8, according to Ensembl DB. Genes of that family are implicated in a number of cellular processes involving alteration
of RNA secondary structure such as translation initiation, nuclear and mitochondrial splicing, and ribosome and spliceosome assembly. Based on their distribution patterns, some members of this family
are believed to be involved in embryogenesis, spermatogenesis, and cellular growth and division.
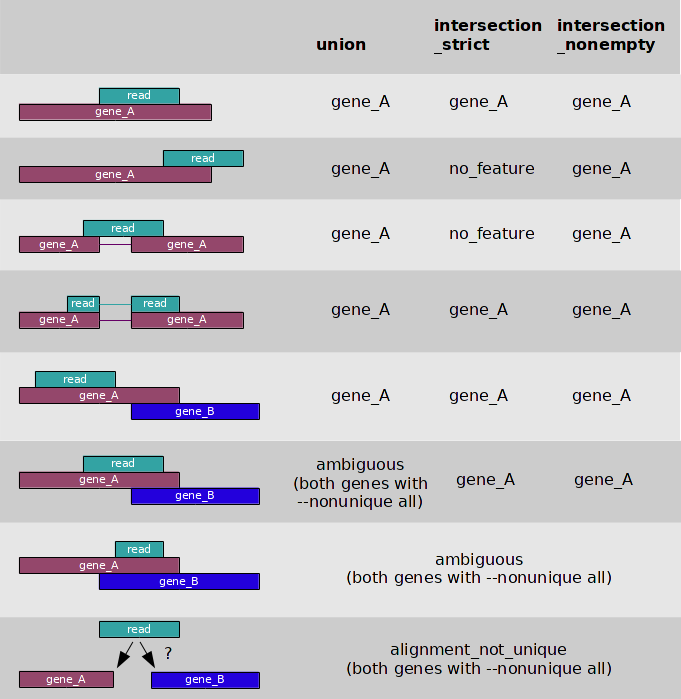
To validate, whether all of the 11 reads are placed exactly in the gene borders, it was decided to run htseq-count with the intersection-strict mode. That run could check the hypothesis, because,
if the read mapped on a gene partly, that run would give a no-feature result (according to a picture presented below). And it seems that all of the 11 reads were mapped exactly into gene coordinates.