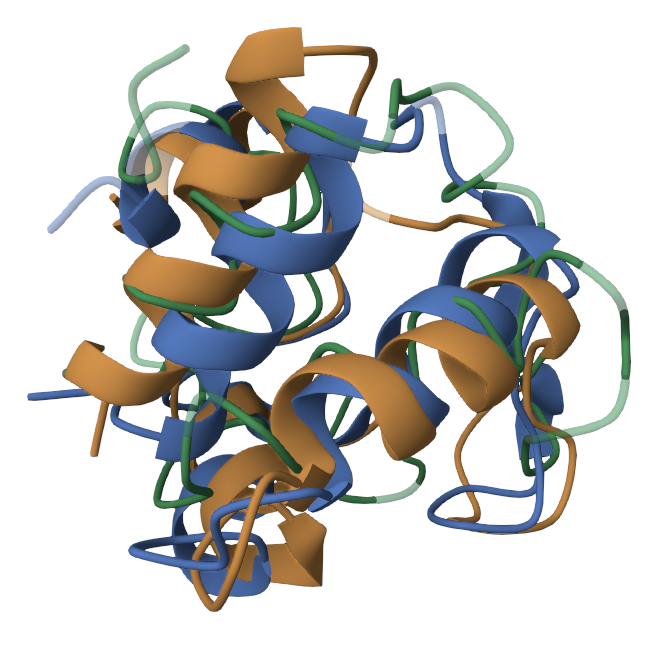
Рис.1 Совмещение структур
1A56 - оранжевый
1COR - синий
1C53 - зеленый
1A56 - оранжевый
1COR - синий
1C53 - зеленый
Cценарий bratz.py (ссылка) запускается с командной строки:
Команда для получения краткой справки по использованию:
Сценарий сравнивает два выравнивания последовательностей. Входные данные — два файла в формате .sto/.stk
Основные функции:
print_help() выводит справку с описанием обязательных параметров и опций:read_alignment(filepath) считывает выравнивание из файла, игнорирует пустые строки и комментарии. Извлекает имя и последовательность, сортирует их по имени.
build_column_vectors(sequences) строит векторы для каждой колонки выравнивания, где каждому символу (кроме дефиса) присваивается номер, а дефису — -.
find_matching_columns(vec1, vec2) находит совпадающие колонки между двумя выравниваниями. Если совпадение найдено, оно добавляется в список.
find_blocks(matches, include_single=False) собирает совпавшие колонки в блоки. Если колонки идут подряд, это считается одним блоком. При опции -i учитываются блоки из 1 колонки.
main() выполняет:Пример:
python bratz.py file1.sto file2.sto -i -o output.txtЭтот запуск учтет блоки длиной в 1 колонку и сохранит результат в output.txt.
Для поиса референсного выравнивания можно обратиться к базе данных BAliBASE (Benchmark Alignment dataBASE) – наиболее признанной. Из архива BAliBASE_R1-5.tar.gz обращаюсь к Reference 5 (RV50), так как инструкции сказанно, что там есть рефересы со внутренними вставками. Рассматриваю файл с вида BBnnn.msf (чтобы открыть в Jalview и в дальнейшем преобразовать в нужный мне формат). Выбрала выравнивание BB50013 (файл), содержащее 18 последовательностей. Белки относятся к метилтрансферазам(EC: 2.1), общий домен thymidylate synthase family.
С референсным выравниванием сравнивались 2 программы из Jalview: ClustalO и Mafft. Для сравнения использовала bratz сценарий (см.выше).
| BAliBASE | ClustalO | ||
|---|---|---|---|
| Длина выравнивания | 386 | 382 | |
| Процент совпадающих колонок | 64.25 | 64.92 | |
| Номер блока | Координаты 1 | Координаты 2 | Длина блока |
| 1 | 1-11 | 1-11 | 11 |
| 2 | 52-67 | 48-63 | 16 |
| 3 | 81-95 | 77-91 | 15 |
| 4 | 110-112 | 105-107 | 3 |
| 5 | 117-122 | 112-117 | 6 |
| 6 | 126-156 | 121-151 | 31 |
| 7 | 159-166 | 154-161 | 8 |
| 8 | 169-172 | 164-167 | 4 |
| 9 | 177-184 | 172-179 | 8 |
| 10 | 195-196 | 190-191 | 2 |
| 11 | 201-227 | 196-222 | 27 |
| 12 | 237-251 | 232-246 | 15 |
| 13 | 255-281 | 250-276 | 27 |
| 14 | 293-296 | 288-291 | 4 |
| 15 | 300-329 | 295-324 | 30 |
| 16 | 337-342 | 332-337 | 6 |
| 17 | 347-353 | 343-349 | 7 |
| 18 | 359-386 | 355-382 | 28 |
| BAliBASE | Mafft | ||
|---|---|---|---|
| Длина выравнивания | 386 | 401 | |
| Процент совдающих колонок | 51.04 | 49.13 | |
| Номер блока | Координаты 1 | Координаты 2 | Длина блока |
| 1 | 1-12 | 1-12 | 12 |
| 2 | 31-33 | 47-49 | 3 |
| 3 | 59-67 | 64-72 | 9 |
| 4 | 88-95 | 93-100 | 8 |
| 5 | 112-112 | 116-116 | 1 |
| 6 | 117-121 | 123-127 | 5 |
| 7 | 136-166 | 142-172 | 31 |
| 8 | 178-184 | 185-191 | 7 |
| 9 | 194-197 | 201-204 | 4 |
| 10 | 201-226 | 208-233 | 26 |
| 11 | 237-250 | 245-258 | 14 |
| 12 | 257-279 | 265-287 | 23 |
| 13 | 295-299 | 303-307 | 5 |
| 14 | 301-320 | 309-328 | 20 |
| 15 | 331-331 | 346-346 | 1 |
| 16 | 347-354 | 362-369 | 8 |
| 17 | 359-371 | 374-386 | 13 |
| 18 | 380-386 | 395-401 | 7 |
Выравнивания проводились с помощью Jalview (ссылка на проект), также есть координаты совпавших колонок для обеих программ: ClustalO и Mafft
Из таблицы видно в целом, что все программы сработали неплохо, так как процент совпадения Mafft около 50%, а в остальных случаях даже выше. Удивительно, что результат у СlustalO оказался немного выше референсного, которое мы принимаем как абсолютно точное. Однако длина выравнивание не слишком большая, поэтому в других случаях показатель скорее всего был бы немного ниже. В обоих выравниваниях встречаются похожие основные высококонсервативные участки гомологии (76-92, 179-187, 202-219, 241-249, 259-289, 314-322). Программы одинаково выровняли участок с длинными инделями (126-166), которые вероятно появились из-за многократной локальной дупликации 6 последовательностей, также можно отметить участок 1-12. В пределах домена общая картина выравниваний в целом сходна. Подобный анализ совпадения блоков во всех программах с разным алгоритмом позволяет сделать выводы о корректности и правильности выравнивания на этих совпадающих участках, это один из вариантов увидеть выравнивание, наиболее близкое к эволюционному, на определенном участке у белков.
Безусловно более точной оказалась программа ClustalO (прогрессивное выравнивание) по суммарному вкладу глобальных характеристик (наивысший процент совпадающих колонок – 64.92%, длина выравнивания близка к референсу, высокая средняя длина совпадающих блоков, а также их большое число). Именно эта программа более точно выделила консервативные участки в пределах домена. У Mafft (итеративное) ниже процент совпадения, такое же число совпадающих блоков, но очень мелких по длине (в частности единичных), большая длина выравнивания по сравнению с референсом. Также появилось много гэпов, по сравнению с другими, которые скорее всего являются эволюционно неверными, из-за чего и большая длина выравниваний и низкий процент совпадения.
Для проведения 3D выравнивания было выбрано следующее семеййство доменов из Pfam: Cytochrome c (PF00034). Для выравнивания выбрала три белка:
| Entry | Chain | RMSD | TM-score | Indentity | Aligned Residues | Sequence length | Modeled Residues |
|---|---|---|---|---|---|---|---|
| 1A56 | A | - | - | - | - | 81 | 81 |
| 1COR | A | 2.6 | 0.67 | 58% | 78 | 82 | 82 |
| 1C53 | A | 3 | 0.51 | 22% | 62 | 79 | 79 |
Программа на PDB не способна построить 3D-выравнивание сразу для всех трех, поэтому экспортирую данные в формате fasta и преобразую в нужный мне вид (файл). В Jalview открывю 3D-выравнивание выравнивание и делаю новое с помощью ClustalO. Теперь их можно будет сравнивать по моему алгоритму (ссылка на проект). Также был создан файл с совпадающими координатами. .
| 3D(PDB) | ClustalO | ||
|---|---|---|---|
| Длина выравнивания | 93 | 87 | |
| Процент совпадающих колонок | 31.18 | 33.33 | |
| Номер блока | Координаты 1 | Координаты 2 | Длина блока |
| 1 | 1-2 | 1-2 | 2 |
| 1 | 6-7 | 5-6 | 2 |
| 3 | 13-18 | 12-17 | 6 |
| 4 | 34-36 | 29-31 | 3 |
| 5 | 38-38 | 33-33 | 1 |
| 6 | 68-68 | 62-62 | 1 |
| 7 | 80-93 | 74-87 | 14 |
При рассмотрении множественного выравнивания наблюдается мало консерватинвых участков: 12-19, 34-57, 72-83 (ClustalO), но с учетом их размера и небольшой длины последовательностей, это очень неплохо. Идентичность 1СOR референсу высока, поэтому имеются хорошо совмещенные вторичные структуры на 3D-выравнивании. У 1C53 многие позиции не являются консервативными и соответствуют совершенно несовместимым многочисленными петлям и изгибам между вторичными структурами (Рис.1). По всей видимости образование разных петель и изгибов с разной топологией и геометрией потребовалось для правильной ориентации сайтов и участков домена Цитохрома С. Совпадающие блоки имеются, однако большой процент с длиной 1. Процент совпадения у СlustalO вновь выше, длина меньше, так как выравнивание этой программой сократило число гэпов. Тем не менее считается, что 3D структуры белков консервативнее их последовательностей.
Clustal - одна из самых широко используемых компьютерных программ для множественного выравнивания нуклеотидных и аминокислотных последовательностей, созданная в 1988 году Десмондом Джерардом Хиггинсом. Последняя версия программы носит название ClustalΩ.
Clustal Omega — это версия, полностью переписанная и обновлённая в 2011 году, написана на языках С и C++ . Она может работать с очень большим количеством (десятки тысяч) последовательностей ДНК/РНК или белков благодаря использованию алгоритма mBed для расчёта направляющих деревьев. Этот алгоритм позволяет очень быстро решать задачи по выравниванию очень больших последовательностей даже на персональных компьютерах. Точность программы была значительно повышена по сравнению с более ранними программами Clustal за счёт использования метода HHalign для выравнивания скрытых марковских моделей профилей. В настоящее время программа используется из командной строки или может быть запущена онлайн.
Программа принимает на вход (нужно как минимум 3 последовательности) широкий спектр форматов: NBRF/PIR, FASTA, EMBL/Swiss-Prot, Clustal, GCC/MSF, GCG9 RSF и GDE. Формат выходного файла может быть следующим: Clustal, NBRF/PIR, GCG/MSF, PHYLIP, GDE, NEXUS.