Задание 1. Получение последовательностей и выравнивание
Для построения филогенетического дерева были взяты последовательности цитохрома B
тех же 11 видов млекопитающих, что и в предыдущей работе.
Был создан файл cyb.list со списком идентификаторов.
Последовательности получены командой seqret из пакета EMBOSS,
выравнивание построено программой MUSCLE:
#получение последовательностей seqret @cyb.list cyb.fasta #волшебное выравнивание muscle -align cyb.fasta -output cyb-alignment.fasta
Длина выравнивания составила 384 позиции, доля гэпов у каждой последовательности не превысила 1,30% (максимум 5 гэпов из 384). Это свидетельствует о высоком качестве выравнивания — цитохром B является консервативным митохондриальным белком.
Задание 2. Конвертация в формат phylip-relaxed
Программа FastME принимает выравнивания только в формате phylip-relaxed.
Для конвертации из FASTA был написан (списан из презентации) скрипт на Python с использованием библиотеки BioPython:
from Bio import AlignIO
inh = open("cyb-alignment.fasta", "r")
outh = open("cyb.phy", "w")
alignment = AlignIO.parse(inh, "fasta")
AlignIO.write(alignment, outh, "phylip-relaxed")
inh.close()
outh.close()
После запуска python3 convert.py создался файл cyb.phy.
Первая строка содержит число последовательностей и длину выравнивания (11 и 384),
каждая следующая — имя последовательности и её аминокислотный состав.
Задание 3. Реконструкция дерева программой FastME
Дерево строилось двумя способами — с моделью p-distance и MtREV.
Описание использованных опций:
-
-i cyb.phy— входной файл с выравниванием в формате phylip-relaxed. -
-pp— флаг-pуказывает на белковые последовательности, букваpзадаёт модель расстояний p-distance — долю различающихся позиций между двумя последовательностями без поправок на множественные замены. -
-pM— флаг-p— белковые данные, букваMзадаёт модель MtREV (Mitochondrial REVersible model) — специальную матрицу замен, разработанную для митохондриальных белков. Более реалистична биологически по сравнению с p-distance (в теории). -
-o <файл>— имя выходного файла с деревом в формате Newick.
#p-distance fastme -i cyb.phy -pp -o cyb_fastme_p.nwk #MtREV fastme -i cyb.phy -pM -o cyb_fastme_mtrev.nwk
Задание 4. Реконструкция дерева программой IQ-Tree
IQ-Tree строит дерево методом максимального правдоподобия
и автоматически подбирает наилучшую модель замен.
iqtree -s cyb.phy
Из созданных программой файлов использовался cyb.phy.treefile —
финальное дерево в формате Newick.
Все три дерева визуализированы в сервисе iTOL
и укоренены в ветвь, наиболее близкую к правильной (нефакт) аутгруппе согласно дереву видов.
Задание 5. Дерево видов
Дерево видов построено в предыдущей работе на основе таксономии NCBI. Давайте восхитимся им еще раз (нет).
Скобочная запись (формат Newick):
Корень дерева делит все виды на две крупные клады — Laurasiatheria: (SORCI, SORCO) и (PIPKU, MYOMA), то есть землеройки и рукокрылые; и Euarchontoglires: ((PSAVE, HYSAF), (LEPYA, SYLPA)) и ((LEMCA, PONPY), TUPGL) - грызуны, зайцеобразные, приматы и тупайи.
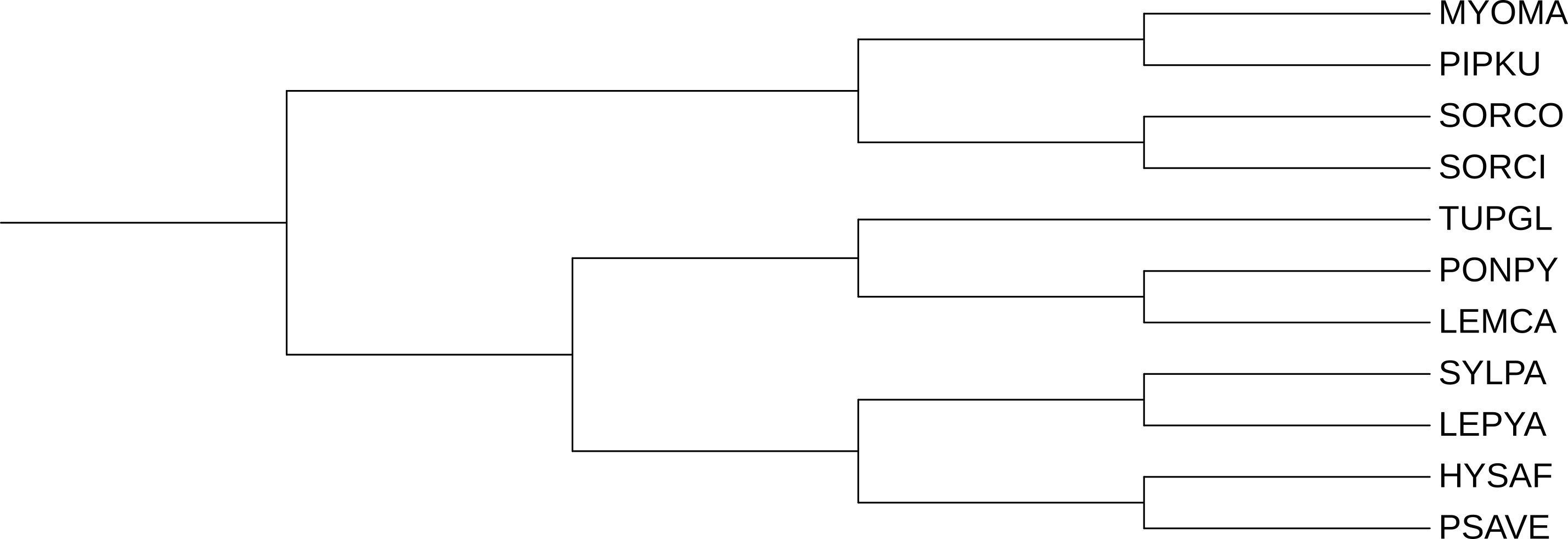
Задание 6. Сравнение дерева видов с деревом FastME (p-distance)
Ниже представлено сравнение дерева, полученного с помощью программы FastME
и модели p-distance, с деревом, основанным на таксономии NCBI.
Дерево укоренено в ветвь, объединяющую MYOMA, PIPKU, TUPGL, SORCI и SORCO —
наиболее близкую к правильной аутгруппе из доступных в данной топологии.

Видно, что ошибки реконструкции следующие:
- Вид TUPGL стал сестринским к кладе (SORCI, SORCO), создав ошибочную кладу (TUPGL, (SORCI, SORCO)). По таксономии TUPGL (Tupaia glis, тупайя) относится к Euarchontoglires и должна быть сестринской к кладе приматов (PONPY, LEMCA).
- Клада (PONPY, LEMCA) стала сестринской к кладе (LEPYA, SYLPA), хотя должна входить в кладу (TUPGL, (PONPY, LEMCA)), которая является сестринской к кладе ((HYSAF, PSAVE), (LEPYA, SYLPA)). Из-за «ухода» TUPGL приматы лишились своего ближайшего соседа по таксономии.
- Из-за вида TUPGL нарушена структура двух глобальных клад, тех, что отходят от корня на дереве таксономии (эти две клады соответствуют Эуархонтоглирам и Лавразиатериям), т.е вклинилась в "лауразиатерийную" группу, разрушив обе клады одновременно.
Клады, восстановленные верно: (SORCI, SORCO), (MYOMA, PIPKU), (PONPY, LEMCA), (LEPYA, SYLPA) — все пары близкородственных видов реконструированы правильно. Ошибки касаются только более глубоких ветвлений.
Задание 7. Сравнение дерева видов с деревом FastME (MtREV)
Далее приведено сравнение дерева, построенного той же программой FastME,
но уже с моделью MtREV, с тем же таксономическим деревом.
Дерево укоренено в ветвь, объединяющую TUPGL, SORCI, SORCO, HYSAF, MYOMA и PIPKU.

Ошибки реконструкции здесь следующие:
- Образовалась ошибочная клада (TUPGL, (SORCI, SORCO)). Как и в предыдущем случае, TUPGL оказалась среди лауразиатерийных видов вместо правильного положения рядом с приматами.
- Образовалась ошибочная клада (HYSAF, (MYOMA, PIPKU)). Вид HYSAF (Hystrix africaeaustralis, дикобраз) относится к Rodentia в составе Euarchontoglires и должен образовывать кладу с PSAVE, однако объединился с рукокрылыми (Chiroptera, Laurasiatheria).
- Вид PSAVE стал сестринским к кладе приматов (PONPY, LEMCA), хотя должен входить в кладу (PSAVE, HYSAF), которая ближе к кладе (LEPYA, SYLPA). Из-за «ухода» HYSAF к рукокрылым PSAVE остался без своего таксономического партнёра.
- Из-за двух видов HYSAF и TUPGL вновь нарушена структура двух глобальных клад. Ни Laurasiatheria, ни Euarchontoglires не восстановлены в правильном составе.
Клады, восстановленные верно: те же четыре пары — (SORCI, SORCO), (MYOMA, PIPKU), (PONPY, LEMCA), (LEPYA, SYLPA). Модель MtREV, несмотря на бо́льшую биологическую реалистичность, не исправила ошибку с TUPGL и добавила новую с HYSAF.
Задание 8. Сравнение дерева видов с деревом IQ-Tree
Наконец, сравнивается дерево, построенное программой IQ-Tree,
с таксономическим деревом.
Дерево укоренено в ветвь, объединяющую PSAVE, TUPGL, SORCI, SORCO, MYOMA и PIPKU.

Ошибки реконструкции следующие:
- Образовалась ложная клада (PSAVE, TUPGL), которая стала сестринской к кладе (SORCI, SORCO), хотя оба этих вида должны быть в другой глобальной кладе — Euarchontoglires. TUPGL должна быть сестринской к приматам, а PSAVE — образовывать кладу с HYSAF.
- Вид HYSAF стал сестринским к кладе (PONPY, LEMCA), хотя вместе с видом PSAVE должен образовывать кладу, сестринскую к кладе (LEPYA, SYLPA). Поскольку PSAVE «ушёл» к TUPGL, HYSAF остался без партнёра и оказался рядом с приматами.
- Вновь глобальное нарушение двух клад, на этот раз из-за видов TUPGL и PSAVE. Оба вида покинули Euarchontoglires и оказались рядом с Laurasiatheria, разрушив структуру обеих глобальных клад, отходящих от корня.
Клады, восстановленные верно: (SORCI, SORCO), (MYOMA, PIPKU), (PONPY, LEMCA), (LEPYA, SYLPA). Метод максимального правдоподобия не справился с правильным размещением TUPGL и PSAVE лучше, чем дистанционные методы.
Задание 9. Итоговое сравнение методов
Все три метода реконструкции одинаково успешно восстановили клады на уровне
близкородственных пар видов: (SORCI, SORCO), (MYOMA, PIPKU),
(PONPY, LEMCA) и (LEPYA, SYLPA). Однако ни один метод не смог верно
разрешить положение TUPGL, HYSAF и PSAVE — видов,
у которых цитохром B эволюционировал с нетипичной скоростью.
Это классический пример эффекта притяжения длинных ветвей (long branch attraction):
алгоритмы группируют быстро эволюционирующие виды вместе независимо от их истинного
таксономического положения, что приводит к нарушению обеих глобальных клад —
Euarchontoglires и Laurasiatheria.
Если сравнивать методы между собой, то FastME с моделью p-distance допустил
наименьшее число ошибок — не на своём месте оказалась только TUPGL.
FastME с моделью MtREV добавил ещё одну ошибку — неверное положение HYSAF,
что неожиданно, поскольку MtREV теоретически более реалистична для митохондриальных
белков. IQ-Tree, несмотря на использование метода максимального правдоподобия,
также допустил две ошибки — неверное положение TUPGL и PSAVE.
Таким образом, в данном конкретном случае ни один из более сложных методов
не превзошёл простую p-distance. Вероятно, проблема не в выборе метода,
а в самом маркере: один ген цитохрома B содержит недостаточно филогенетического
сигнала для надёжного разрешения глубоких ветвлений при данном наборе таксонов.
Для получения более точного результата вероятно стоит использовать несколько
независимых генов или полные митохондриальные геномы.
Назад