Нуклеотидный BLAST
-
Ген, кодирующий δ-субъединицу АТФ-синтазы
Для выполнения последующих двух исследований и анализов необходимо было получить некоторые данные о гене δ-субъединицы АТФ-синтазы для выбранного мною в прошлом практикуме организма (это S. cerevisiae). Из файлов, скачанных и представленных в прошлом практикуме, были получена следующая необходимая информация о данном гене у данного организма:
NP_010280.1 – идентификатор δ-субъединицы, ее аминокислотную последовательность можно посмотреть в fasta файле
NC_001136.10 – идентификатор нуклеотидной записи, к которой относится данный ген (оказалось это хромосома 4). Для его получения был использован первый идентификатор.
neighbourhood_of_gene– фаста-файл, содержащий участок с координатами 442.762..443.776 их четвертой хромосомы, данный участок содержит исследуемый ген 443.029..443.511, ниже представлено изображение данного участка (Рис. 1) (данные получены благодаря второму идентификатору):
 Рис.1 Изображение участка ДНК, содержащего данный ген
Рис.1 Изображение участка ДНК, содержащего данный ген
- BLAST
АТФ-синтаза – один из самых известных и распространенных белков среди аэробных организмов. Мне захотелось проверить его консервативность на примере структуры гена и конкретного белка, входящего в состав АТФ-синтазы (собственно δ-субъединицы моего ранее выбранного организма).
Для проведения данного исследования необходимо было выбрать таксон, достаточно далекий от исходного организма (поскольку интересует именно консервативность глобально, а не среди родственников), поскольку мой организм не принадлежит ни к первичноротым, ни к вторичноротым животным, то таксон я решила выбирать среди первых. Методом тыка остановаилась на Araneae (taxid:6893). Поиск нужной информации происходился с помощью двух типов программы BLAST (об этом ниже) на сайте. База данных для поиска была взята довольно известная база данных RefSeq Genome Database, в которой было 4 геномных сборок, входящих в этот таксон. Программы были следующие:
Программа blastn – для начало захотелось проверить насколько сильно могут быть похожи именно гены (а точнее даже экзоны) данной субъединицы, поэтому и использовалась эта программа (для поиска небольших похожих нуклеотидных последовательностей среди неблизкородственных организмов). На вход подавался фрагмент из файла n_gene.fasta (описанный выше), а именно интересующий меня ген (в программе указывались параметры from и to, координаты гена в участке известны, если перевести их на счет с 1, то будет 267-749). Параметры были взяты по умолчанию (E-value = 10, word_size = 11). Результатов не нашлось. Алгоритм требует найти в геноме паука непрерывный участок из 11 нуклеотидов, идеально совпадающий с геном из дрожжей. Вероятность того, что даже у консервативного гена сохранится идеальный 11-нуклеотидное "слово" чрезвычайно мала. Мутации(синонимичные/несинонимические замены; вставки, делеции, изменения в некодирующих регионах (интронах)) гарантированно "испортят" такое короткое точное совпадение. Также на вход добавлось лишь 4 генома, что очень мало для поиска совпадений.
Программа tblastn – здесь уже в качестве запроса подавалась уже сама последовательность белка (смотри fasta-файл в самом первом абзаце), параметры были взяты также по умолчанию (E-value = 0.05, word_size = 5), данный алгоритм нужен для сравнения белка-запроса с транслированными последовательностями из базы данных, результаты можно увидеть в файле. Как видно из результатов, обнаружилось 4 находок, среди всех представителей семейства. Можно наблюдать довольно высокий процент покрытия для локального выравнивания (30-32%) и довольно высокий процент идентичности (в среднем 50%). на Рис. 2 можно видеть, какие участки наиболее оказались схожи у дрожжей и пауков. На основании этого, а также крайне низкого значения E-value, можно действительно убедиться, что в общем-то данный белок является высококонсервативным, по крайней мере его последовательность не так уж и сильно отличается между такими разными и далекими организмами.
 Рис.2 Наиболее похожие участки белка
Рис.2 Наиболее похожие участки белка
- Поиск в геноме эукариота генов основных рибосомальных РНК по далекому гомологу
В этом задании я попыталась отыскать гомологов 16S рРНК (последовательность) и 23S рРНК (последовательность) E. coli у дрожжей. Для этого сперва был проиндексирован мой геном (файл .fna) следующей командой:
makeblastdb -in GCF_000146045.2_R64_genomic.fna -dbtype nucl
После чего был запущен алгоритм blastn (был выбран именно он, так как он подходит для поиска похожих некодирующих белки нуклеотидных последовательностей даже среди неблизкородственных организмов), в качестве запроса были поданы как раз выше упомянутые последовательности 16S и 23S рРНК. Команды следующие:
blastn -task blastn -query 16srna.fna -db GCF_000146045.2_R64_genomic.fna -out res16_2 -outfmt 7
blastn -task blastn -query 23srna.fna -db GCF_000146045.2_R64_genomic.fna -out res23_2 -outfmt 7Результаты выдачи blastn можно посмотреть здесь: 16S - таблица и текст, 23S - таблица и текст. (текстовый формат был получен аналогичными командами, но без -outfmt 7). Запросы дали 9 и 25 находок соответственно.
Малая субъединица рибосомы содержит одну длинную молекулу рРНК, которая является её структурным и функциональным ядром. У бактерий (E. coli) это 16S рРНК. У эукариот (как дрожжи) в цитоплазматических рибосомах это 18S рРНК. У эукариот в митохондриальных рибосомах это 15S-18S рРНК (у дрожжей — 15S). Значит когда ищем гомолог бактериальной 16S рРНК в эукариотическом организме, по умолчанию ищем 18S рРНК (ядерная) или 15S рРНК (митохондриальная).
В результате работы blastn были найдены следующие гомологи гена 16s рРНК Escherichia coli: два гена 18s рРНК на хромосоме XII и ген 15s рРНК на мтДНК. Ещё одна находка соответствует гену гипотетический белка на хромосоме VII.
Гены 18S рРНК на хромосоме XII - это как раз те два лучших "попадания" в NC_001144.5 (хромосома XII) с Score ~56.3 и E-value = 4e-07. В геноме дрожжей рибосомные гены организованы в тандемный повтор. Это значит, что одна и та же последовательность, кодирующая предшественник рРНК (которая потом нарезается на 18S, 5.8S и 25S рРНК), многократно повторяется друг за другом. BLAST нашел два таких повтора на хромосоме XII. Первый ген: расположен на минус-цепи. Координаты ~455936 - 455978. Второй ген: Также на минус-цепли. Координаты ~465073 - 465115
Ген 15S рРНК на митохондриальной ДНК - Это NC_001224.1 (митохондриальный геном) с Score = 54.5 и E-value = 1e-06. Ген ориентирован на плюс-цепь. BLAST показывает выравнивания в районе координат ~7489-7595 и ~8128-8167.
"Ген гипотетического белка" на хромосоме VII - Это находка с низким score (NC_001139.9, E-value = 4.5) — скорее всего, случайное совпадение. E-value = 4.5 означает, что вы могли бы ожидать около 4-5 таких случайных выравниваний просто по воле случая при поиске в базе данных такого размера. Это совпадение не является статистически значимым и не представляет собой реальный гомолог. Его можно смело игнорировать.
Для гена 23s рРНК Escherichia coli были найдены ген 21s рРНК на мтДНК, два гена 25s рРНК на хромосоме XII. Остальные находки либо накладываются на аннотированные белки, либо соответствуют неаннотированным участкам генома.Ген 21S рРНК на митохондриальной ДНК: Так же, как и в случае с 16S/15S рРНК, 21S рРНК является гомологом бактериальной 23S рРНК в составе митохондриальной рибосомы дрожжей. Высокий Score (109 bits) и чрезвычайно низкий E-value (4e-23). Это указание на гомологию. Длинные выравнивания (137/187 и 123/171 нуклеотидов) с высокой идентичностью (~73%). Это консервативные участки, отвечающие за ключевые функции рибосомы.
Гены 25S рРНК на хромосоме XII: В цитоплазматической рибосоме эукариот гомологом бактериальной 23S рРНК является 25S/28S рРНК (у дрожжей — 25S). Высокий Score (74.3 bits) и очень низкий E-value (3e-12). Результат статистически значим. Множественные попадания в одни и те же локусы хромосомы XII (позиции ~452210, ~461347, ~490178). Это как раз и есть проявление тандемных повторов гена рРНК. BLAST нашел один и тот же консервативный участок в нескольких соседних копиях этого повтора.
Остальные находки (Хромосомы XVI, IV, XIII, VI, II): E-value = 8.6 буквально означает, что при поиске в базе данных такого размера можно случайно найти около 8 таких же выравниваний. Это статистически незначимо. Короткие выравнивания (20-25 нуклеотидов).
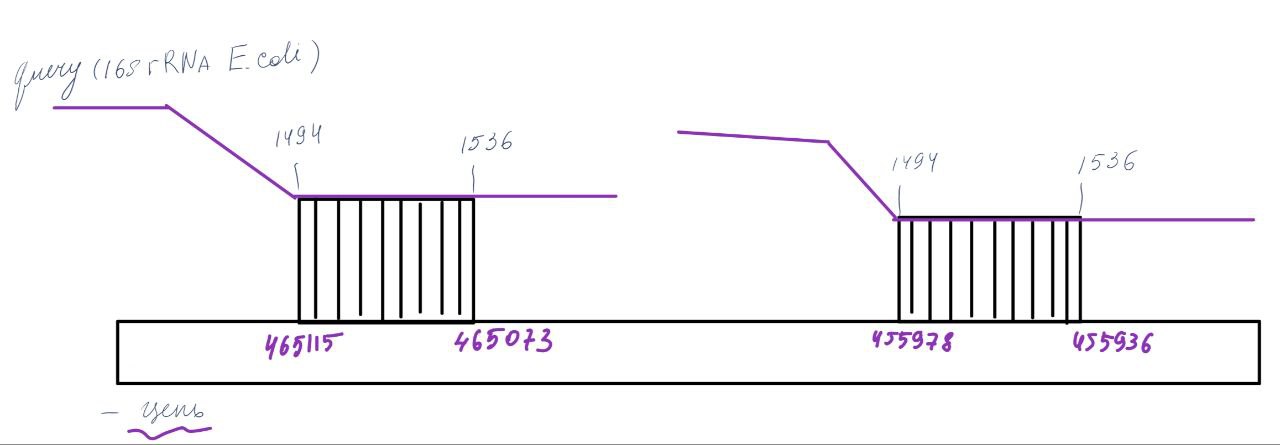 Рис.3 Схематичное изображение результатов на примере двух находок
Рис.3 Схематичное изображение результатов на примере двух находок
- Карты локального сходства
Для выполнения данного задания поиск необходимых нуклеотидных последовательностей осуществлялся на сайте базы данных ENA с помощью следующего запроса:
tax_tree(1578) AND description="chromosome"
Отобрались следующие две последовательности для сравнения: хромосома Lactobacillus paragasseri (нуклеотидный AC CP054573.1) и хромосома Lactobacillus kefiranofaciens subsp. kefirgranum (нуклеотидный AC CP098405.1). Таким образом сравнивались вид и подвид другого вида, принадлежащих роду Lactobacillus. Сравнение происходило выдачи двух разных алгоритмов blast для данных последовательностей, а именно сравнивались выдача megablast, blastn, tblastx (параметры во всех случаях были взяты по умолчанию). Отбор последовательностей необходимо было произвести так, чтобы эти три выдачи отличались. Увы, только две программы построили карты локального сходства.
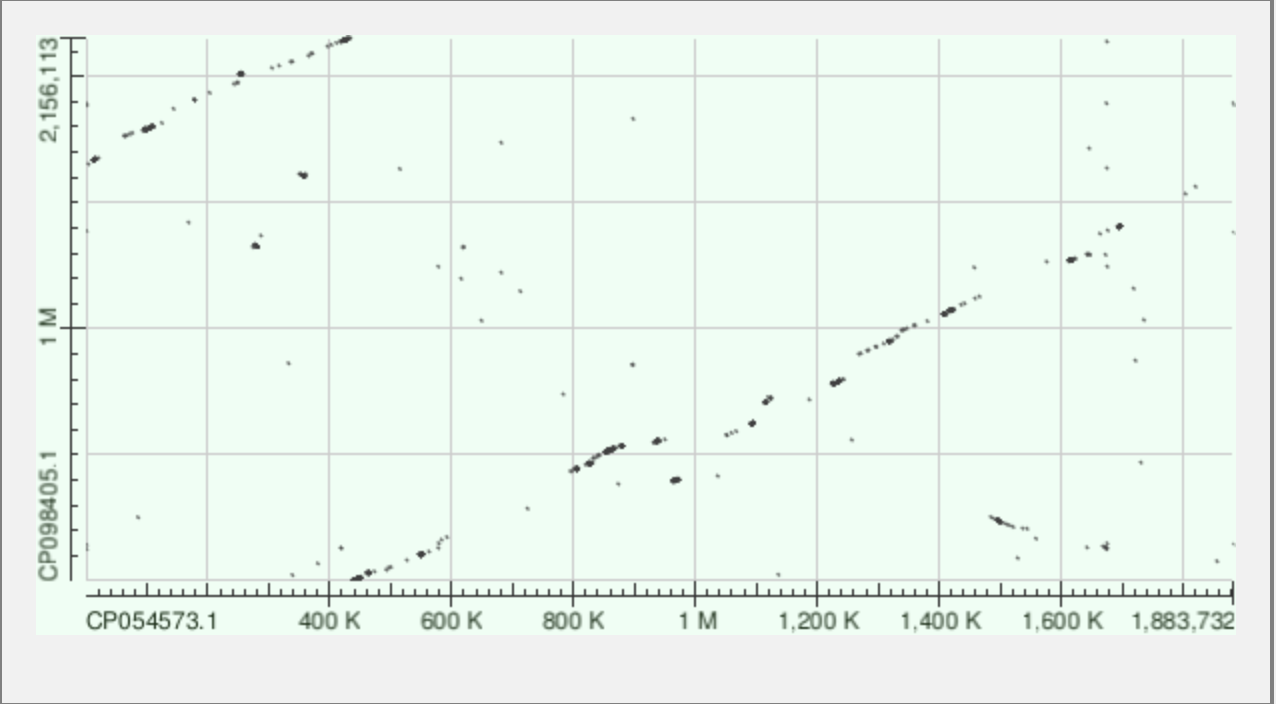 Рис.4 выдача megablast
Рис.4 выдача megablast
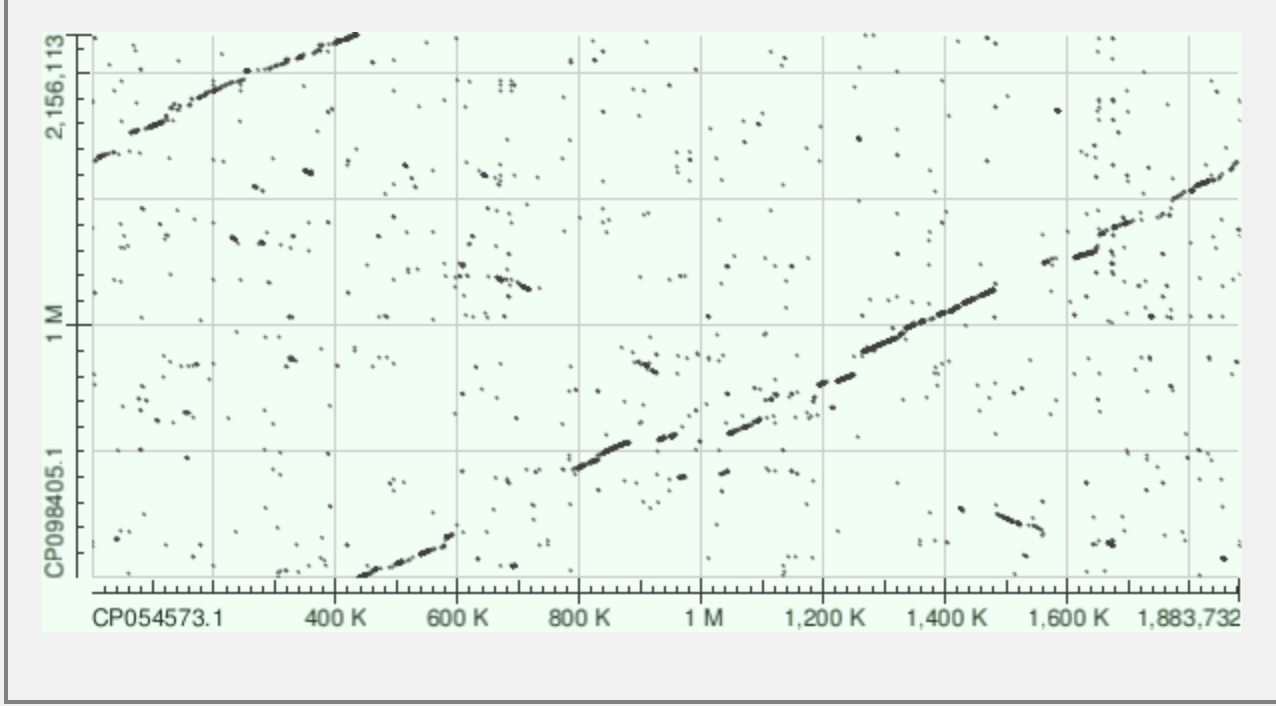 Рис.5 выдача blastn
Рис.5 выдача blastnКак видно из Рис. 4 и 5, выдача megablast не слишком информативна (по ней не скажешь о схожести этих двух последовательностей и родстве бактерий, также на ней не видны различия в геномах, о которых написано ниже, это и неудивительно, так как данный алгоритм хорошо подходит только для почти полностью идентичных последовательностей), выдача же blastn гораздо более содержательна, по ней далее и будет проведен анализ и сравнение геномов.
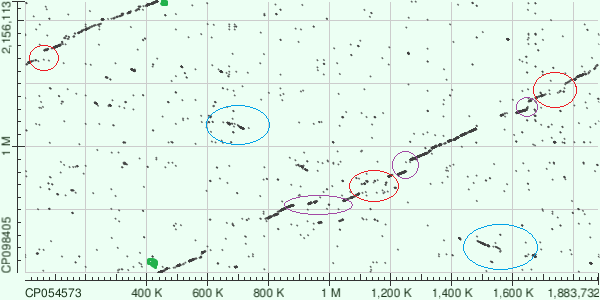 Рис.6 Описание карты
Рис.6 Описание карты
- Глобально можно заметить, что в обоих случаях был выбран один и тот же тип цепи, то есть данные цепи не комплементарны (об этом общий ход прямой на графике).
- Зеленой точкой на карте отмечен разрыв, это говорит о том, что в качестве начала для записи последовательности были выбраны разные точки на кольцевых хромосомах.
- Красным отмечены неконсервативные участки, то есть те, где по всей видимости последовательности крайне слабо похожи, или вовсе эти зоны соответствуют инделям и гэпам в выравнивании, об этом можно сделать вывод на основании того, что в данных зонах как будто бы пропущен участок линии на графике.
- Фиолетовым отмечены зоны инсерций/делеций, в первом случае (самый левый овал) либо произошла крупная вставка в нижней последовательности, либо крупная делеция в боковой, во втором и третьем случаях соответственно произошли либо небольшие вставки в боковой последовательности, либо небольшие делеции в нижней. Об этом можно судить по скачкам линий (по их разрывам и нарушении непрерывности в соответствующих участках).
- Голубым же цветом можно наблюдать транспозицию выделенных участков, которая приэтом сопряжена с инверсией в обоих случаях (наклон линии в другую сторону).
- В целом также можно наблюдать на карте большое число мелких точек и набросков, они соответствуют многочисленным повторам в последовательностях.
В итоге можно заметить, что несмотря на то, что организмы таксономически довольно близки (по сути это просто разные виды одного рода), в их геномах можно обнаружить довольно глобальные отличия и перестройки: делеции, инсерции, повторы, транспозиции и даже инверсии. По всей видимости это связано с конкретными видовыми особенностями данных организмов.
- BLAST