Практикум 12:
Множественное выравнивание последовательностей белков, сравнение пар выравниваний.
Для выполнения заданий практикума 12 было использовано семейство Pfam исследованное в рамках практикума 11. В качестве программ для выравнивания применялись алгоритмы Clustal Omega, Muscle, Mafft, MSAprobs. При помощи VerAlign в первом случае сравнивались выравнивания полученные при помощи Clustal Omega и Muscle. Во втором Mafft и MSAprobs.
SP score: 0.77 (доля одинаково выровненных колонок в тестовом и референсном выравниваниях) CS score: 0.34 (доля одинаково выровненных позиций) avg_SPdist score: 0.93 (среднее расстояние между двумя несовмещёнными колонками по всем колонкам референсного выравнивания) Наибольшее несовпадение наблюдается между позициями 27 и 41, в неконсервативном участке, посчитано в ручную.
SP score: 0.86 CS score: 0.25 avg_SPdist score: 0.95
Наибольшее несовпадение пришлось на тот неконсервативный участок, в позициях 28-42, а также на участок 54-58 посчитано в ручную.
Структурное выравнивание и его сравнение с алгоритмом MSA.

В качестве объекта для структурного выравнивания я решил выбрать альфа амилазу трёх хордовых животных, а именно японской рисовой рыбы(3VM5), человека(1С8Q) и свиньи(1BVN).
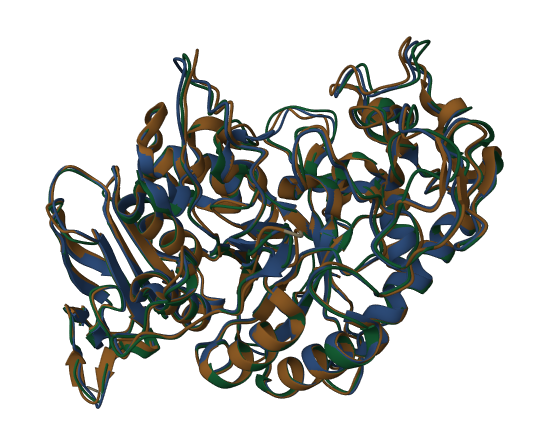
В качестве референса была выбрана структра альфа-амилазы рисовой рыбы, поскольку альфа-амилаза человека и свиньи слишком близки по структуре. Как можно видеть на рисунке структуры всех трёх белков обладают очень схожей структурой и обладают внушительным процентом идентичных остатков, что свидетельствует об очень небольшом эволюционном расхождении. Результаты структурного выравнивания совпадают с результатами полученными в результате множественного выравнивания MSA.
Описание программы MUSCLE.
Программа множественного выравнивания белковых и нуклеотидных последовательностей. Алгоритм состоит из трёх этапов: Эскизный прогрессивный- На этом первом этапе алгоритм производит множественное выравнивание, делая упор на скорость, а не на точность. Этот шаг начинается с вычисления k-мерного расстояния для каждой пары входных последовательностей для создания матрицы расстояний. UPGMA кластеризует матрицу расстояний для создания двоичного дерева. Из этого дерева строится прогрессивное выравнивание, начиная с создания профилей для каждого листа дерева. Для каждого узла в дереве строится попарное выравнивание двух дочерних профилей, создавая новый профиль, который будет назначен этому узлу. Это продолжается до тех пор, пока не произойдет выравнивание всех входных последовательностей в корне дерева. Улучшенный прогрессивный-На этом этапе основное внимание уделяется получению более оптимального дерева путем вычисления "расстояния Кимуры" для каждой пары входных последовательностей с использованием выравнивания нескольких последовательностей, полученного на первом этапе, и создается вторая матрица расстояний. UPGMA кластеризует эту матрицу расстояний, чтобы получить второе двоичное дерево. Прогрессивное выравнивание выполняется для получения множественного выравнивания последовательностей, как на этапе 1, но оно оптимизируется только путем вычисления выравнивания в поддеревьях, порядок ветвления которых изменился по сравнению с первым двоичным деревом, что приводит к более точному выравниванию. Этап проверки-На этом заключительном этапе ребро выбирается из второго дерева, при этом края посещаются на уменьшающемся расстоянии от корня. Выбранное ребро удаляется, разделяя дерево на два поддерева. Затем вычисляется профиль множественного выравнивания для каждого поддерева. Новое выравнивание нескольких последовательностей создается путем повторного выравнивания профилей поддерева. Если оценка SP улучшается, новое выравнивание сохраняется, в противном случае оно отбрасывается. Процесс удаления ребра и выравнивания повторяется до тех пор, пока не произойдет сходимость или пока не будет достигнут заданный пользователем предел.