Ресеквенирование. Поиск и описание полиморфизмов у человека.
Часть I: Подготовка чтений
Нам были даны файлы с одноконцевыми чтениями экзома человека участка хромосомы.
Моей была хромосома 6.
Все файлы из таблиц ниже можно найти тут: /nfs/srv/databases/ngs/catherine.nesterenko
| Входной файл |
Выходной файл |
Команда |
Описание |
| chr6.fastq |
chr6_fastqc.zip |
fastqc chr6.fastq |
Программой FastQC была получена информация о качестве чтений. |
| chr6.fastq |
trim.fastq |
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.
jar SE -phred33 chr6.fastq trim.fastq TRAILING:20 MINLEN:50 |
Программой Trimmomatic были убраны концы прочтений, у которых качество ниже 20, и убраны прочтения меньше
50 нуклеотидов длинной |
| trim.fastq |
trim_fastqc.zip |
fastqc trim.fastq |
Программой FastQC была получена информация о качестве редактированных чтений. |
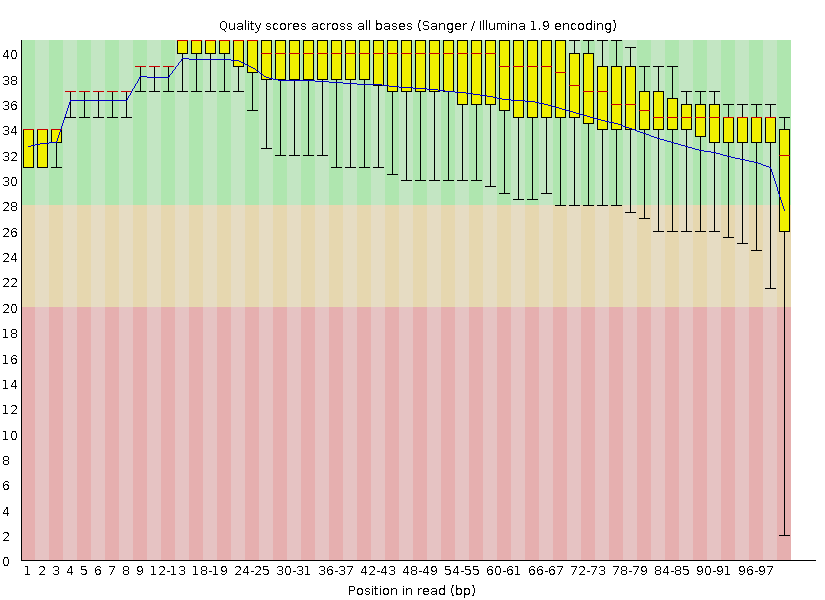
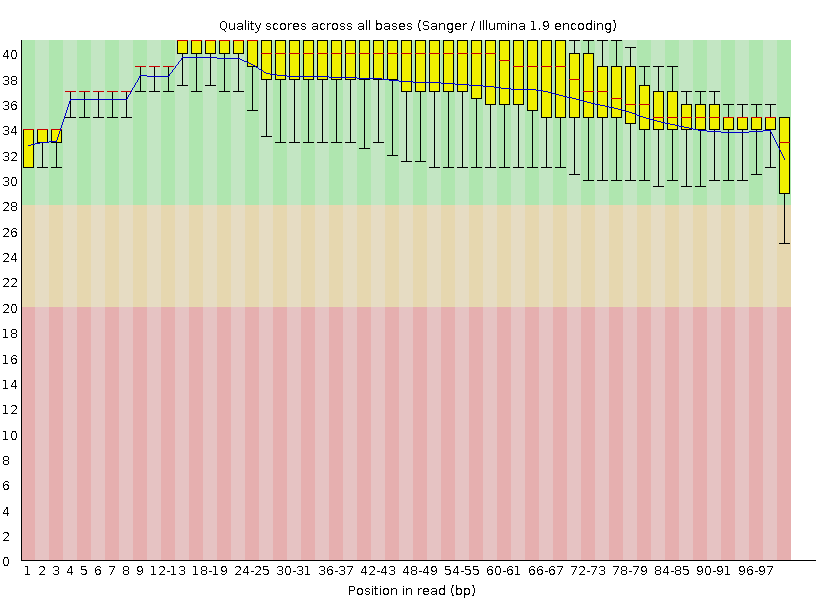
Качество прочтений (Per base sequence quality)
|
|
|
До использования Trimmonatic
|
После использования программы Trimmonatic
|
Число чтений до чистки: 10289
Число чтений после чистки: 10123
Количество ридов уменьшилось на 166, потому что были отсеяны риды с качеством прочтения ниже
20 (Q < 20) и риды длиной менее 50 нуклеотидов. На картинках ниже это представленно наглядно - пропали риды с
длинными усами. Качество прочтения вычисляется по формуле Q=10*lg(p), p - вероятность ошибки.
Часть II: Картирование чтений
| Входной файл |
Выходной файл |
Команда |
Описание |
| ch6.fasta |
ch6-ind.x.ht2, где x от 1 до 8 |
hisat2-build chr6.fasta ch6-ind |
Индексируем референсную последовательнось. |
| ch6-ind.x.ht2, где x от 1 до 8 |
alinch.sam |
hisat2 -x ch6-ind -U trim.fastq --no-softclip
--no-spliced-alignment -S alinch.sam |
Построение выраванивания прочтений и референса в формате .sam. |
| alinch.sam |
balinch.bam |
samtools view alinch.sam -b -o balinch.bam |
Изменение формата .sam в его бинарный аналог - .bam. |
| balinch.bam |
balinch.sorted.bam |
samtools sort balinch.bam balinch.sorted |
Сортировка выравнивания чтений с референсом по координате в референсе начала чтения. |
| balinch.sorted.bam |
balinch.sorted.bam.bai |
samtools index balinch.sorted.bam |
Индексируем отсортированный файл. |
Картировалось на хромосому 10046 ридов. 77 ридов не картировалось совсем.
Программа Hisat2 дает такую информацию о выраванивании чтений на геном, как:
координата, куда "легло" чтение, количество картирований для конкретного чтения, некую
оценку качества выравнивания и другие характеристики.
Часть III: Анализ SNP
| Входной файл |
Выходной файл |
Команда |
Описание |
| balinch.sorted.bam, chr6.fasta |
polym.bcf |
samtools mpileup -uf chr6.fasta balinch.sorted.bam -o polym.bcf |
Создание файла с полиморфизмами. |
| polym.bcf |
diff.vcf |
bcftools call -cv polym.bcf -o diff.vcf |
Поиск отличий между референсом и чтениями. |
Описание трех полморфизмов
| № |
Координаты |
Тип полиморфизма |
В референсе |
В ридах |
Глубина покрытия |
Качество чтений |
| 1 |
107006115 |
Делеция |
TCCCCC |
TCCCC,TCCC |
5 |
15,1077 |
| 2 |
106978314 |
Замена |
G |
A |
170 |
183,009 |
| 3 |
107016838 |
Инсерция |
CTTT |
CTTTTT |
36 |
217,468 |
Всего полиморфизмов найдено - 84. Из них 5 инделей.
Покрытие неравномерное, как и качество. Наибольшая глубина покрытия 170, наименьшая 1.
Аннотация SNP
Необходимо аннотировать полученые полифорфизмы. Будем работать с SNP и использовать такие базы данных,
как refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
| Команда |
Описание |
convert2annovar.pl -format vcf4 diff.vcf -outfile ch6.avinput |
Изменение формата файла с полиморфизмами для работы с annovar. |
annotate_variation.pl -out refgen-an -build hg19 -dbtype refGene
ch6.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация в Refgene. |
annotate_variation.pl -filter -out db-an -build hg19 -dbtype
snp138 ch6.avinput /nfs/srv/databases/annovar/humandb.old |
Аннотация в dbnsp. |
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19
-out 1000G ch6.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация в 1000genomes. |
annotate_variation.pl -regionanno -build hg19 -out GWAS
-dbtype gwasCatalog ch6.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация в GWAS. |
annotate_variation.pl ch6.avinput /nfs/srv/databases/annovar/humandb.old/
-filter -dbtype clinvar_20150629 -buildver hg19 -out CLINV |
Аннотация в CLINVAR. |
База данных refseq в annovar делит snp на группы по позиции в геноме. На экзонах - 15,
на интронах - 64, 3'UTR - 5. Эти данные можно получить из файла refgen-an.variant_function.
В файле refgen-an.exonic_variant_function можно найти информацию о делении snp в экзонах
на группы - синонимичные (3), несинонимичные (10) и те, что привели к нонсенс мутации (2).
Гены, на которые попали snp - AIM1,TNFAIP3, OPRM1.
У 73 snp есть rs.
Заболевания, к которым могут привести snp, можно найти в файле GWAS.hg19_gwasCatalog.
Аннотированные в GWAS заболевания.
| Инсульт |
106987370 106987370 |
A -> C |
| Системная красная волчанка |
138195723 138195723 |
C -> G |
| Ревматоидный артрит. Системная красная волчанка |
138196066 138196066 |
T -> G |
| Ишемическая болезнь сердца |
154414563 154414563 |
A -> G |
Аннотация в ClinVar выдала одну запись в файле. На 138196066 позиции в геноме замена T -> G. Она описана
термином 'not specified', это говорит, что она либо благоприятная, либо безвредная мутация, либо является
признаком для еще не определенного заболевания.
Аннотация в 1000 genomes дала информацию о частоте встречаймости той или иной замены. В файл 1000G.hg19_ALL.sites.2014_10_dropped
попало 73 записи.
| Частота замены |
Координата в геноме |
Замена |
| Наибольшая |
0.987819 |
107008422 |
C -> T |
| Наименьшая |
0.00658946 |
154382572 |
G -> T |
Ссылка на сводную таблицу в Excel.
© Нестеренко Екатерина 2018