Анализ транскриптомов
Анализ качества чтений
Для анализа даны прочтения, полученные в результате секвинирования РНК. Все указанные файлы хранятся
тут: /nfs/srv/databases/ngs/catherine.nesterenko/pr12
| Входной файл |
Выходной файл |
Команда |
Описание |
| chr6.1.fastq |
chr6.1_fastqc.zip |
fastqc chr6.1.fastq |
Программой FastQC была получена информация о качестве чтений. |
| chr6.1.fastq |
chr6trim.fastq |
java -jar
/nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.
jar SE -phred33 chr6.1.fastq chr6trim.fastq TRAILING:20 MINLEN:45 |
Программой Trimmomatic были убраны концы прочтений, у которых качество ниже 20, и убраны прочтения меньше
45 нуклеотидов длинной |
| chr6trim.fastq |
chr6trim_fastqc.zip |
fastqc chr6trim.fastq |
Программой FastQC была получена информация о качестве редактированных чтений. |
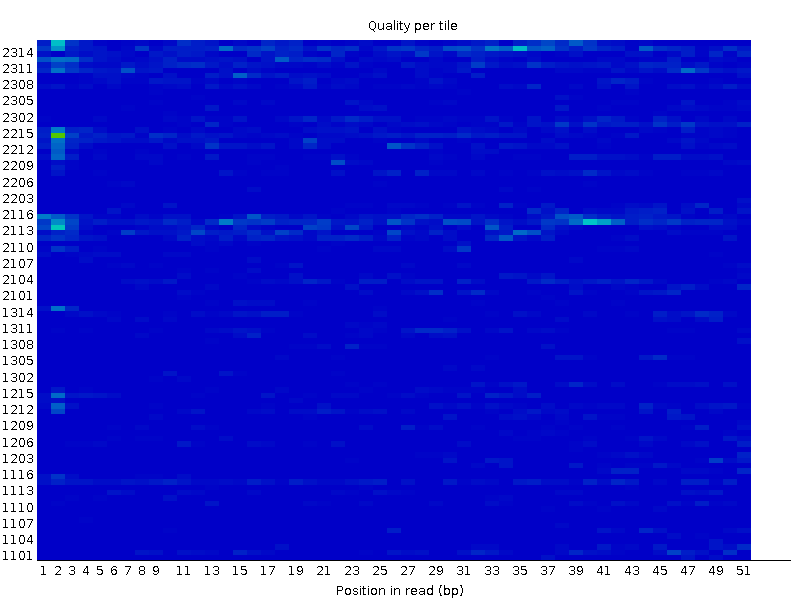
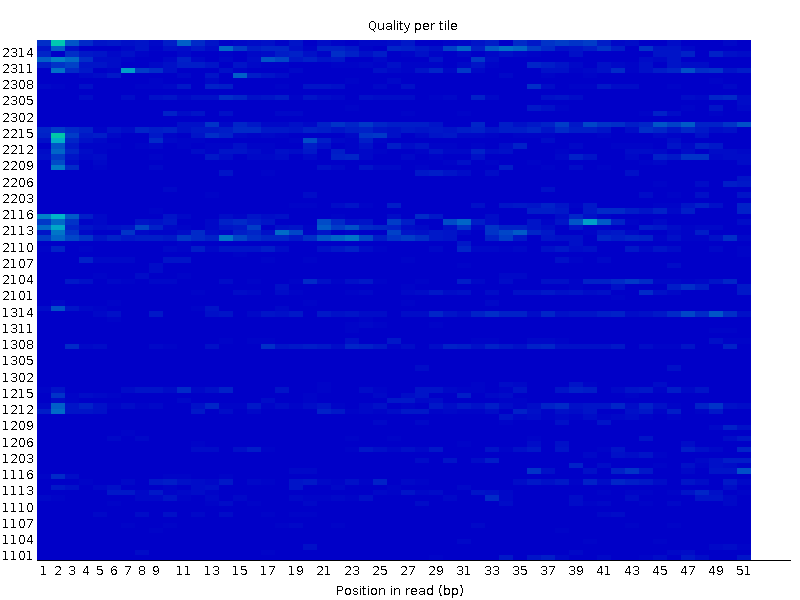
Качество прочтений (Per tile sequence quality)
|
|
|
До использования Trimmonatic
|
После использования программы Trimmonatic
|
Диаграмма с 'усами' была не информативна, так как все риды были довольно хорошего качества. Ограничение
длины рида 45 помогает улучшить показатель Per tile sequence quality. Теплые тона показывают нуклеотиды
плохого качества, ниже среднего. На фотографиях видно, как меняется качество после работы Trimmomatic.
Картирование чтений и анализ выравнивания
| Входной файл |
Выходной файл |
Команда |
Описание |
| ch6.fasta |
ch6-ind.x.ht2, где x от 1 до 8 |
hisat2-build chr6.fasta ch6-ind |
Будем использовать проиндексированный в прошлом практикуме референс. Команда для работы из
директории прошлого практикума. |
| ch6-ind.x.ht2, где x от 1 до 8 |
al.sam |
hisat2 -x ../ch6-ind -U ch6trim.fastq --no-softclip -S al.sam |
Построение выраванивания прочтений и референса в формате .sam. Убираем опцию
--no-spliced-alignment, так как выравниваем РНК с ДНК, где есть интронные участки. |
| al.sam |
al.bam |
samtools view al.sam -b -o al.bam |
Изменение формата .sam в его бинарный аналог - .bam. |
| al.bam |
sorted.bam |
samtools sort al.bam sorted |
Сортировка выравнивания чтений с референсом по координате в референсе начала чтения. |
| sorted.bam |
sorted.bam.bai |
samtools index sorted.bam |
Индексируем отсортированный файл. |
Картировалось на хромосому 44761 ридов. 9234 ридов не картировалось совсем.
Программа Hisat2 дает такую информацию о выраванивании чтений на геном, как:
координата, куда "легло" чтение, количество картирований для конкретного чтения, некую
оценку качества выравнивания и другие характеристики.
Подсчет чтений
Для подсчета чтений использовалась команда htseq-count с некоторыми опциями.
Документация к htseq-count.
- -f - формат файла с вырвниванием (.sam или .bam) Работаем с.bam.
- -s - {yes,no,reverse}, параметр, показывающий взята ли информация из цепь-специфичного анализа. Yes -
по умолчанию. Берем no, так как не знаем на какой цепи закодированы наши последовательности.
- -i - GFF атрибут, используется как feature-ID. Для последовательностей с РНК возьмем gene_id.
- -m - {union,intersection-strict,intersection-nonempty}, режим для обработки тех ридов, которые легли с
перекрытиями или на более, чем одну features. Используем параметр intersection-nonempty, чтобы учитывать
любое перекрывание.
| Входной файл |
Выходной файл |
Команда |
Описание |
| sorted.bam |
h-count.txt h1-count.txt |
htseq-count -f bam -s no -m intersection-nonempty -i
gene_id sorted.bam /P/y14/term3/block4/SNP/rnaseq_reads/
gencode.v19.chr_patch_hapl_scaff.annotation.gtf >> h-count.txt |
Подсчет количества чтений, описание параметров можно увидеть выше.
Запущен поиск был два раза, чтобы посмотреть на разницу.
Второй раз с опциями -s и -m по умолчанию. |
| h-count.txt, h1-count.txt |
1itog.txt, 2itog.txt |
grep -w -v 0 h-count.txt >> 1itog.txt |
Используем команду для быстрого извлечения результатов. |
Итоги, выдача файлов:
| 1itog.txt |
2itog.txt |
ENSG00000156508.13 44586
__no_feature 175
__not_aligned 9234
|
ENSG00000156508.13 19
__no_feature 44742
__not_aligned 9234
|
Почему такая большая разница в количестве ридов? Потому, что в первом случае мы разрешаем любым,
хоть как-нибудь упавшим ридам на данный ген засчитываться. А во втором случае по умолчанию у опциии -m
параметр unique. И его рекомендуется использовать в большинстве случаев, так сказано в документации. (см. выше)
С его использованием засчитываются те риды, которые хоть как-то уникально упали на ген. То есть,
если рид полностью лежит внутри гена Х и частично внутри гена У, то программа с этим параметром не засчитает его,
как рид, упавший на хоть какой-нибудь ген. Данному риду будет присвоена метка __ambiguous. Опция -s, ее наличие
или отсутствие, не влияет на количество посчитанных ридов.
Не все риды легли точно в границы гена. Риды, видимо, не могли лечь на соседние экзонные участки,
так как в первом случае запуска htseq-count должно было бы быть больше генов, на которые что-то да упало. Довольно
большое количество ридов вообще не выровнялось с хромосомой.
Риды легли на один ген - ENSG00000156508.13. (EEF1A1, eukaryotic translation elongation factor 1 alpha 1).
Этот ген кодирует изоформу альфа-субъединицы комплекса фактор элонгации-1, которая отвечает за ферментативную доставку аминоацильных тРНК в рибосому.
Изоформа (альфа 1) экспрессируется в мозге, плаценте, легких, печени, почках и поджелудочной железе.
Было обнаружено, что этот ген имеет несколько копий на многих хромосомах, некоторые из которых, если не все,
представляют разные псевдогены.
© Нестеренко Екатерина 2018